Sigmatropisk reaktion: Definition, mekanisme og eksempler (Cope & Claisen)
Sigmatropisk reaktion: klar definition, mekanisme og eksempler (Cope & Claisen). Lær om [3,3]-omlægninger, pericykliske processer og praktiske anvendelser.
En sigmatropisk reaktion i organisk kemi er en pericyklisk reaktion. En sigmatropisk reaktion anvender ikke en katalysator og involverer et enkelt molekyle (en ikke-katalyseret intramolekylær proces). Den ændrer en σ-binding til en anden σ-binding. Navnet sigmatropisk er resultatet af en sammensætning af den længe etablerede betegnelse "sigma" for enkelte kulstof-kulstof-bindinger og det græske ord tropos, der betyder drejning. Der er tale om en omarrangeringsreaktion, hvilket betyder, at bindingerne i et molekyle forskydes mellem atomer, uden at der udgår atomer fra eller tilføjes nye atomer til molekylet. I en sigmatropisk reaktion flytter en substituent sig fra en del af et π-bundet system til en anden del i en intramolekylær reaktion med samtidig omlægning af π-systemet. Ægte sigmatropiske reaktioner kræver normalt ikke en katalysator. Nogle sigmatropiske reaktioner katalyseres af en Lewis-syre. Sigmatropiske reaktioner har ofte overgangsmetalkatalysatorer, der danner mellemprodukter i analoge reaktioner. De mest kendte af de sigmatrope omlægninger er [3,3] Cope-omlægningen, Claisen-omlægningen, Carroll-omlægningen og Fischer-indolsyntesen.
Billedgalleri
10 Billeder









Notation og mekanistisk oversigt
Sigmatropiske omlægninger betegnes ofte som [i,j]-reaktioner, hvor tallene i og j angiver antallet af atomer i hvert af de to π-systemer, målt fra den oprindelige σ-binding. For eksempel er en [3,3]-omlægning en process hvor σ-bondens endepunkter ender tre positioner fra hinanden i hver af de to deltagende π-systemer. Disse reaktioner er typisk pericykliske og foregår via et enkelt, concerted overgangsforløb uden frie radikaler eller ioner (medmindre en særlig variant eller katalyse indfører sådanne intermediater).
Elektronisk og stereokemisk styres sigmatropiske reaktioner af Woodward–Hoffmann-reglerne: termiske [i,j]-sigmatropiske reaktioner foregår typisk suprafacialt (samme side af π-systemet) for de almindelige lave-order tilfælde ([1,5], [3,3] osv.), mens photokemisk aktivering kan ændre den tilladte symmetri og dermed give andre stereokemiske udfald. Geometriske begrænsninger (fx ringstørrelse) kan gøre en antarafacial migrationsvej nødvendig, men sådanne veje er ofte vanskelige at gennemføre i praksis.
Cope-omlægningen (klassisk og varianter)
Cope-omlægningen er en prototypisk [3,3]-sigmatropisk omlejring af 1,5-dien-systemer. Den involverer omlejring af bindinger i et 1,5-dien til isomeriske 1,5-dien-produkter. Nogle vigtige punkter:
- Cope-omlægningen kan være degenereret, dvs. produktet er identisk med udgangsmolekylet, så reaktionen er i ligevægt.
- Substituenter påvirker termodynamikken: f.eks. gør 3,3-dialkylsubstitution (Claisen-strain relief og hyperkonjugation) ofte omlægningen termodynamisk favoriseret.
- Oxy-Cope og anionisk oxy-Cope: hvis der er en hydroxylgruppe på passende position, kan oxy-Cope-followed-by-tautomerisering give en karbonylforbindelse (en enon eller aldehyd). Den anioniske variant, hvor hydroxylgruppen først deprotoneres, sænker aktiveringsenergien markant og gør omlægningen meget mere letdreven ved milde betingelser.
- Overgangsstatetypisk er chair-lignende (lavere energi) eller boat-lignende afhængigt af substituenternes krav, hvilket bestemmer stereokemi i produktet.
Claisen-omlægningen og dens varianter
Claisen-omlægningen er også en [3,3]-sigmatropisk reaktion, klassisk for allyl-ethers (f.eks. allyl vinyl-ethers, allyl aryl-ethers). Ved en typisk Claisen-omlægning dannes en ny C–C-binding, og produkter inkluderer allylerede karbonylforbindelser eller o-allylphenoler fra allylphenyl-ethers. Nogle varianter og vigtige punkter:
- Der findes flere tilpasninger af Claisen: Ireland–Claisen (gennem ester-enolat intermediater), Johnson–Claisen (alkoksycarbonyl varianter), Eschenmoser–Claisen (iminium-baserede varianter) osv., som udvider anvendeligheden under mildere betingelser eller med kontrol over stereokemi.
- Claisen-omlægningen er nyttig i syntese for at flytte allylgrupper og bygge op konjugerede systemer og funktionaliserede carbonylforbindelser.
- Ligesom for Cope spiller overgangsstatets geometri (chair vs. boat) og substituenternes placering en afgørende rolle for stereokemien i produktet.
Stereokemi og Woodward–Hoffmann-reglerne
Woodward–Hoffmann-symmetrireglerne for pericykliske reaktioner forklarer, hvilke sigmatropiske omlægninger der er tilladte under termiske forhold og hvilke der kræver fotonisk aktivering. For de almindelige [3,3]-omlægninger betyder det:
- Termisk [3,3] → suprafacial-suprafacial overførsel af elektronpar er symmetry-allowed, hvilket sikrer en koncerted proces med bevarelse af stereokemi (kopler cis/trans-forhold i substrat til produkt på en forudsigelig måde).
- Fotokemisk aktivering kan tillade alternativer, fx antarafacial komponenter, men ofte med ændret stereokemisk udfald.
Katalyse, metalkomplekser og påvirkning af reaktionsbetingelser
Selvom mange sigmatropiske reaktioner er termisk drevne uden katalysator, kan både Lewis-syre-katalyse og overgangsmetalkomplekser ændre barriererne eller føre til alternative mekanismer:
- Lewis-syrer kan koordinere til funktionen (fx oxygen) og stabilisere overgangstilstanden, sænke aktiveringsenergien og gøre reaktionen mulig ved lavere temperatur.
- Overgangsmetaller kan interagere med π-systemer og danne π-allyl-intermediater, som effektivt ændrer reaktionsforløbet fra en concerted pericyklisk proces til en stepwise mekanisme — det kan være ønsket for at opnå andre selektiviteter.
- Anioniske varianter (fx anionisk oxy-Cope) initieres ved base-deprotonering og giver ofte dramatisk lavere aktiveringsenergi og hurtigere omlejring.
Anvendelser og praktiske eksempler
Sigmatropiske omlejninger anvendes bredt i organisk syntese:
- Cope og oxy-Cope-omlægninger anvendes til konstruktion af cykliske skeletoner og til at omarrangere konjugerede systemer. Den anioniske oxy-Cope bruges ofte til at frembringe en stabiliseret enolanion, der efterfølgende tautomeriserer til en karbonylforbindelse.
- Claisen-omlægningen er et standardværktøj til at installere allylgrupper ortho til aromatiske oxygen-funktioner (f.eks. allylering af fenoler) eller til opbygning af konjugerede carbonylsystemer; Ireland–Claisen er populær til stereokontrolleret dannelse af γ,δ-umættede carbonylforbindelser.
- Hvor pericykliske reaktioner generelt er stereospecifikke, bruges de ofte i totalsyntese af naturlige produkter for at sikre stereokemisk renhed.
Termodynamik, kinetik og praktiske hensyn
Om en sigmatropisk omlejring går til produkt afhænger af både termodynamik og kinetik. Mange Cope-omlægninger er reversible og kræver termodynamiske faktorer (substituent-effekter, forløsning af ringspænding, efterfølgende tautomerisering) for at fortrænge ligevægten mod produktet. Reaktionsbetingelserne — temperatur, opløsningsmiddel, tilstedeværelse af base eller Lewis-syre — afgør ofte, hvilken variant (termisk, fotokemisk, anionisk eller metalassisteret) man vælger i praktisk syntese.
Kort résumé
Sigmatropiske reaktioner er en central klasse af pericykliske omlejringer i organisk kemi, karakteriseret ved intramolekylær migration af en σ-binding i forbindelse med omlægning af et π-system. De mest anvendte eksempler er [3,3]-omlægninger som Cope og Claisen, som giver kraftfulde og stereospecifikke værktøjer i organisk syntese. Forståelse af pericyklisk mekanisme, Woodward–Hoffmann-symmetri og effekten af substituenter og katalysatorer er afgørende for at kunne udnytte disse reaktioner effektivt.
Oversigt over sigmatropiske forskydninger
Woodward-Hoffman-nomenklatur for sigmatropisk forskydning
Der anvendes en særlig notation til at beskrive sigmatropiske forskydninger. Hvert af kulstofatomerne på molekylets rygrad tildeles et positionsnummer. Sigmatropiske omlægninger beskrives ved hjælp af et ordensterm [i,j]. Dette betyder, at en σ-binding, der støder op til et eller flere π-systemer, vandrer til en ny position (i-1) og (j-1) atomer, der er fjernet fra σ-bindingens oprindelige placering. Når summen af i og j er et lige tal, er dette et tegn på, at der er tale om en neutral kæde af alle C-atomer. Et ulige tal antyder, at der er et ladet C-atom eller et heteroatoms lone pair, der erstatter en kulstof-kulstof-dobbeltbinding. Således bliver [1,5]- og [3,3]-forskydninger til [1,4]- og [2,3]-forskydninger med heteroatomer, samtidig med at symmetriovervejelser bevares. Hydrogener er udeladt i det tredje eksempel af hensyn til klarheden.

Her er en måde at finde rækkefølgen af en given sigmatropisk omlægning på. Det første skridt er at give numre til hvert atom, idet man starter med atomer i den binding, der brydes, som atom 1. Kemikere tæller atomerne i hver retning fra den brudte binding til de atomer, der danner den nye σ-binding i produktet. De tal, der svarer til de atomer, der danner den nye binding, adskilles derefter med et komma og sættes i parentes. Dette giver den sigmatropiske reaktionsordningsdeskriptor.
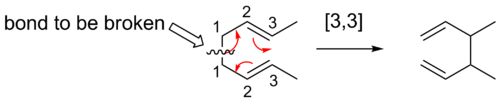
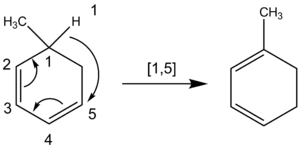
Kemikere tæller også atomer, når de nævner et sigmatropisk skift, hvor et hydrogenatom bevæger sig. Kulstofkæden brydes ikke ved en vandring af et hydrogenatom. Kemikere tæller derfor på tværs af alle de atomer, der er involveret i reaktionen, i stedet for kun på tværs af de nærmeste atomer. F.eks. er følgende hydrogenatomvandring af orden [1,5], som opnås ved at tælle mod uret gennem π-systemet, snarere end [1,3] gennem ringens CH-gruppe, som fejlagtigt ville blive resultatet, hvis man talte 2med uret.
Suprafaciale og antarafaciale forskydninger
Kemikere har undersøgt sigmatropiske reaktioner, hvor den migrerende gruppe har et sterocenter. I princippet kan alle sigmatropiske forskydninger forekomme med enten samme (retention) eller modsat (inversion) geometri for den migrerende gruppe. Dette afhænger af, om det migrerende atoms oprindelige bindingslobe eller dets anden lobe anvendes til at danne den nye binding.
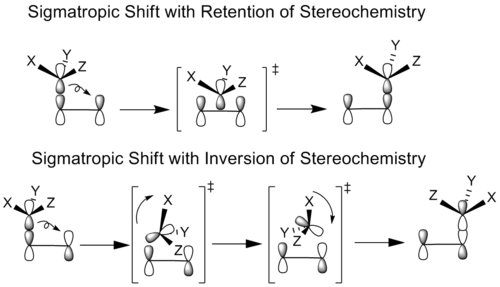
I tilfælde af stereokemisk retention oversætter den migrerende gruppe uden rotation til bindingspositionen. I tilfælde af stereokemisk inversion roterer og translaterer den migrerende gruppe både for at nå sin bundet konformation.
Der er en anden måde, hvorpå en sigmatropisk reaktion kan give produkter med forskellig sterokemi. Den migrerende gruppe kan forblive på π-systemets oprindelige side efter genbinding. Eller den kan gå til den modsatte side af π-systemet. Hvis den migrerende gruppe forbliver på den samme side af π-systemet, kaldes skiftet suprafacialt. Hvis den migrerende gruppe overføres til den modsatte side, kaldes det et antarafacialt skift. Transformationer, der finder sted inden for små eller mellemstore ringe, kan ikke foretage antarafaciale forskydninger.
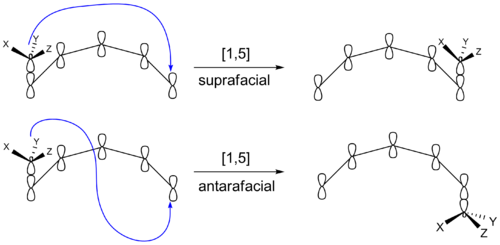
Klasser af sigmatropiske omlægninger
[1,3] Skift
Termiske hydridforskydninger
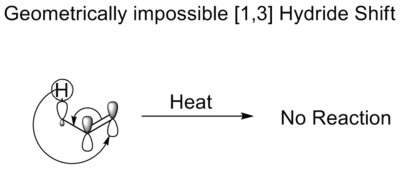
I et termisk [1,3]-hydridskifte flytter et hydrid tre atomer. Woodward-Hoffmann-reglerne dikterer, at det ville foregå i et antarafacial skifte. Selv om et sådant skift er symmetrisk tilladt, forbyder den Mobius-topologi, der kræves i overgangstilstanden, et sådant skift. Det er geometrisk umuligt. Det er derfor, at enoler ikke isomeriseres uden en syre- eller basekatalysator.
Termiske alkylforskydninger
Termiske alkyl [1,3]-forskydninger skal i lighed med [1,3]-hydridforskydninger foregå antarafacielt. Overgangstilstandens geometri er prohibitiv. Men en alkylgruppe kan på grund af dens orbitals natur vende sin geometri og danne en ny binding med den bageste lobe af dens 3sp-orbital. Denne reaktion vil resultere i en suprafacial forskydning. Disse reaktioner er stadig ikke almindelige i åbne kædesystemer på grund af overgangstilstandens meget velordnede natur. Reaktionerne fungerer derfor bedre i cykliske molekyler.
![[1,3] Alkyl Shifts](https://www.alegsaonline.com/image/550px-1%2C3alkylfixed.png)
Fotokemiske [1,3] Skift
Fotokemiske [1,3]-forskydninger burde være suprafaciale forskydninger; de fleste er imidlertid ikke-koncerterede, fordi de foregår gennem en triplettilstand (dvs. de har en diradikal mekanisme, som Woodward-Hoffmann-reglerne ikke gælder for).
[1,5] Skift
Et [1,5]-skift indebærer, at 1 substituent (-H, -R eller -Ar) flyttes nedad 5 atomer i et π-system. Det er blevet vist, at hydrogenet skifter i både cykliske og åbne kædesystemer ved temperaturer på eller over 200 ˚C. Disse reaktioner forudsiges at foregå suprafacielt ved hjælp af en Huckel-topologisk overgangstilstand.
![[1,5] Hydride shift in a cyclic system](https://www.alegsaonline.com/image/300px-1%2C5hydridecyclicfixed.png)
Fotostråling ville kræve en antarafacial forskydning af brint. Selv om sådanne reaktioner er sjældne, er der eksempler på, at antarafaciale forskydninger er foretrukne:
![Antarafacial [1,5] Hydride Shift](https://www.alegsaonline.com/image/600px-1%2C5hantarafacialfixed.png)
I modsætning til hydrogen [1,5]-forskydninger er der aldrig blevet observeret [1,5]-alkyforskydninger i et system med åben kæde. Kemikere har bestemt hastighedspræferencer for [1,5]alkylforskydninger i cykliske systemer: carbonyl og carboxyl> hydrid> phenyl og vinyl>>> alkyl.
Alkylgrupper undergår [1,5]-forskydninger meget dårligt og kræver normalt høje temperaturer. For cyclohexadiener er temperaturen for alkylforskydninger imidlertid ikke meget højere end for carbonylgrupper, som er den bedst migrerende gruppe. En undersøgelse viste, at dette skyldes, at alkylforskydninger på cyclohexadiener foregår gennem en anden mekanisme. Først åbnes ringen, efterfulgt af et [1,7]-skift, og derefter reformeres ringen elektrocyklisk:
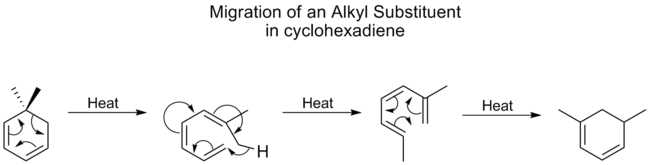
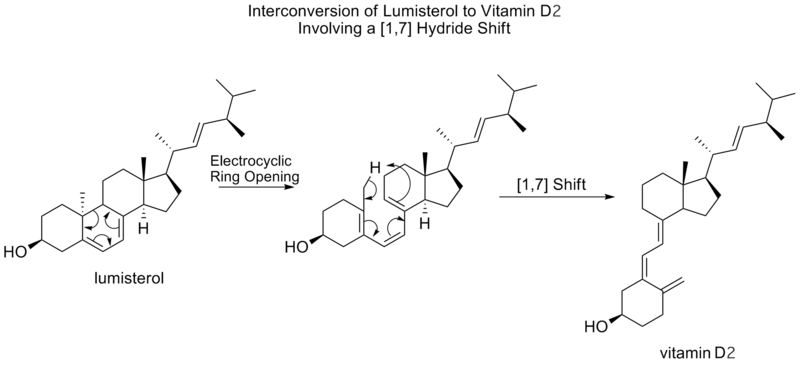
Denne samme mekanistiske proces ses nedenfor, uden den afsluttende elektrocykliske ring-lukningsreaktion, i interkonverteringen af lumisterol til D-vitamin2.
[1,7] Skift
[1,7] sigmatropiske forskydninger forudsiges af Woodward-Hoffmann-reglerne at foregå antarafacialt, ved en Mobius-topologisk overgangstilstand. Et antarafacialt [1,7]-skift er observeret ved omdannelsen af lumisterol til D-vitamin 2, hvor der efter en elektrocyklisk ringåbning til prævitamin D 2sker en methylbrinteskift.
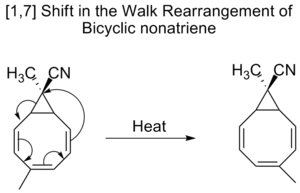
Bicykliske nonatriener undergår også [1,7]-forskydninger i en såkaldt walk-omlægning, som er forskydning af tosidig gruppe, som en del af en treleddet ring, i et bicyklisk molekyle.
[3,3] Skift
[3,3] sigmatropiske forskydninger er velundersøgte sigmatropiske omlægninger. Woodward-Hoffman-reglerne forudsiger, at disse reaktioner med seks elektroner vil foregå suprafacielt ved hjælp af en Huckel-topologisk overgangstilstand.
Claisen-omlægning
Claisen-omlægningen, der blev opdaget i 1912 af Rainer Ludwig Claisen, er det første registrerede eksempel på en [3,3]-sigmatropisk omlægning. Denne omlægning er en nyttig reaktion, der danner kulstof-kulstof-bindinger. Et eksempel på Claisen-omlægning er [3,3]-omlægningen af en allylvinylether, som ved opvarmning giver en γ,δ-umættet carbonyl. Dannelsen af en carbonylgruppe gør denne reaktion, i modsætning til andre sigmatropiske omlejringer, i sagens natur irreversibel.

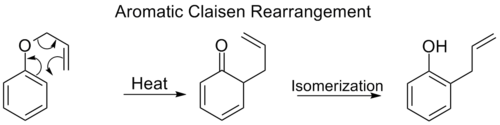
Aromatisk Claisen-omlægning
ortho-Claisen-omlægningen indebærer [3,3]-forskydning af en allylphenylether til et mellemprodukt, som hurtigt tautomeriseres til en ortho-substitueret phenol.
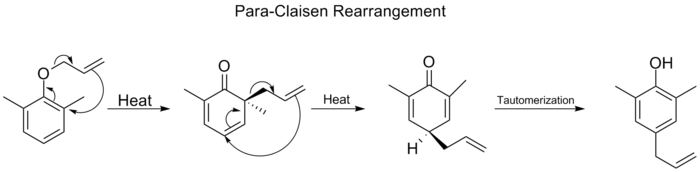
Når begge ortho-positioner på benzenringen er blokeret, vil der ske en anden ortho-Claisen-omlægning. Denne para-Claisen-omlægning ender med tautomerisering til en tri-substitueret phenol.
Omlægning af Cope
Cope-omlægningen er en udførligt undersøgt organisk reaktion, der involverer [3,3]-sigmatropisk omlægning af 1,5-diener. Den blev udviklet af Arthur C. Cope. For eksempel giver 3,4-dimethyl-1,5-hexadien opvarmet til 300 °C 2,6-octadien.

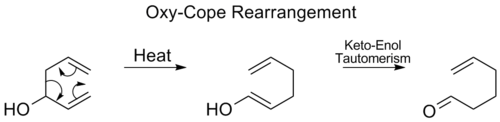
Oxy-Cope-omlægning
Oxy-Cope-omlægningen tilføjes en hydroxylgruppe ved C3 og danner en enal eller enon efter keto-enol-tautomerisme af den mellemliggende enol:
Carroll-omlægning
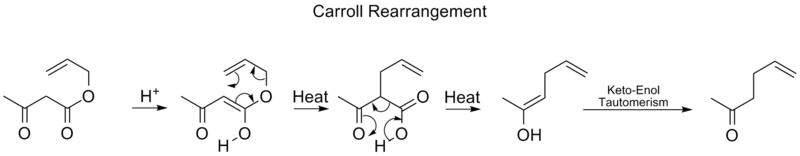
Carroll-omlægningen er en omlægningsreaktion i organisk kemi og indebærer omdannelse af en β-keto allylester til en α-allyl-β-ketocarboxylsyre. Denne organiske reaktion kan efterfølges af decarboxylering, og det endelige produkt er en γ,δ-allylketon. Carroll-omlægningen er en tilpasning af Claisen-omlægningen og er i realiteten en decarboxylativ allylation.
Fischer Indol-syntese
Fischer-indolsyntesen er en kemisk reaktion, der producerer den aromatiske heterocykel indol fra en (substitueret) phenylhydrazin og en aldehyd eller keton under sure forhold. Reaktionen blev opdaget i 1883 af Hermann Emil Fischer.
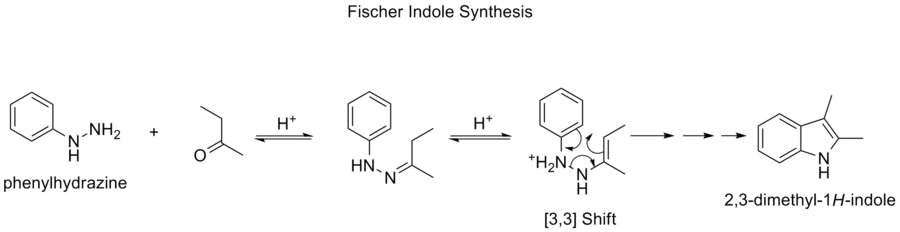
Valget af syrekatalysator er meget vigtigt. Blandt de vellykkede syrekatalysatorer kan nævnes: Bronsted-syrer som HCl, H2SO4 , polyphosphorsyre og p-toluensulfonsyre. Lewis-syrer som bor-trifluorid, zinkchlorid, jernchlorid og aluminiumchlorid er også nyttige katalysatorer.
Der er blevet offentliggjort flere anmeldelser.
[5,5] Skift
I lighed med [3,3]-forskydninger forudsiger Woodward-Hoffman-reglerne, at [5,5]-sigmatropiske forskydninger vil foregå suprafacielt, Huckel-topologiens overgangstilstand. Disse reaktioner er sjældnere end [3,3]-sigmatropiske forskydninger, men dette skyldes hovedsagelig, at molekyler, der kan gennemgå [5,5]-forskydninger, er sjældnere end molekyler, der kan gennemgå [3,3]-forskydninger.
![[5,5] shift of phenyl pentadienyl ether](https://www.alegsaonline.com/image/800px-5%2C5shiftfixeds.png)
Omlægning af gåture
Migrationen af en tosidig gruppe, såsom O, S, NR eller CR2 , som er en del af en treleddet ring i et bicyklisk molekyle, betegnes almindeligvis som en walk-omlægning. Dette kan formelt karakteriseres i henhold til Woodward-Hofmann-reglerne som værende en (1, n) sigmatropisk forskydning. Et eksempel på en sådan omlægning er forskydning af substituenter på tropilidener (1,3,5-cycloheptatriener). Ved opvarmning gennemgår pi-systemet en elektrocyklisk ringlukning og danner bicycle[4,1,0]heptadien (norcaradien). Derefter følger et [1,5]-alkylskifte og en elektrocyklisk ringåbning.
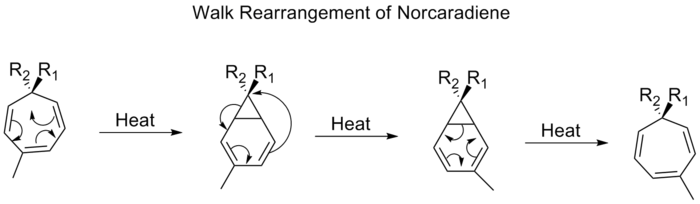
Ved at gå gennem et [1,5]-skifte forventes det, at omlægningen af norcaradiener foregår suprafacielt med bevarelse af stereokemien. Eksperimentelle observationer viser imidlertid, at 1,5-skiftet af norcaradiener foregår antarafacielt. Teoretiske beregninger viste, at [1,5]-skiftet er en diradikal proces, men uden at involvere nogen diradikale minima på den potentielle energioverflade.
Relaterede sider
- 2,3-sigmatropisk omlægning
- NIH-skift
- Grænsepunkts teori om molekylære orbitaler
- Woodward-Hoffmann-regler
Spørgsmål og svar
Q: Hvad er en sigmatropisk reaktion i organisk kemi?
A: En sigmatropisk reaktion er en pericyklisk reaktion, der involverer en ukatalyseret intramolekylær proces, og som ændrer en σ-binding til en anden σ-binding.
Q: Indebærer en sigmatropisk reaktion en katalysator?
A: En egentlig sigmatropisk reaktion involverer normalt ikke en katalysator, selvom nogle sigmatropiske reaktioner kan katalyseres af en Lewis-syre.
Q: Hvad betyder udtrykket "sigmatropisk"?
A: Udtrykket "sigmatropisk" er et sammensat ord, der består af "sigma", som refererer til enkelte kulstof-kulstofbindinger, og det græske ord "tropos", som betyder drejning.
Q: Hvilken slags reaktion er en sigmatropisk reaktion?
A: En sigmatropisk reaktion er en omlejringsreaktion, hvilket betyder, at bindingerne i et molekyle skifter mellem atomer, uden at nogen atomer forlader eller nye atomer føjes til molekylet.
Q: Hvad sker der i en intramolekylær sigmatropisk reaktion?
A: I en intramolekylær sigmatropisk reaktion bevæger en substituent sig fra en del af et π-bundet system til en anden del med samtidig omlejring af π-systemet.
Q: Er der nogle velkendte sigmatropiske omlejringer?
A: Nogle af de mest kendte sigmatropiske omlejringer er [3,3] Cope omlejring, Claisen omlejring, Carroll omlejring og Fischer indol syntese.
Q: Involverer sigmatropiske reaktioner ofte overgangsmetalkatalysatorer?
A: Ja, sigmatropiske reaktioner har ofte overgangsmetalkatalysatorer, der danner mellemprodukter i analoge reaktioner.
Relaterede artikler
Forfatter
AlegsaOnline.com Sigmatropisk reaktion: Definition, mekanisme og eksempler (Cope & Claisen) Leandro Alegsa
URL: https://da.alegsaonline.com/art/90314
Kilder
- dx.doi.org : 10.1021/ja01044a027
- dx.doi.org : 10.1039/C39760000873
- dx.doi.org : 10.1021/ja01034a076
- dx.doi.org : 10.1016/0040-4039(75)80050-5
- dx.doi.org : 10.1002/anie.197208321
- dx.doi.org : 10.1002/cber.19120450348
- dx.doi.org : 10.1002/cber.19250580207
- dx.doi.org : 10.1002/cber.19260590927
- dx.doi.org : 10.1021/ja01859a055
- dx.doi.org : 10.1021/ja00754a042
- dx.doi.org : 10.1021/ja00026a007
- dx.doi.org : 10.1021/ja01076a067
- dx.doi.org : 10.1039/JR9400000704
- dx.doi.org : 10.1002/cber.188301602141
- dx.doi.org : 10.1002/cber.188401701155