Creutzfeldt-Jakobs sygdom (CJD) – dødelig prionsygdom: symptomer og forløb
Creutzfeldt-Jakobs sygdom (CJD): dødelig prionsygdom. Læs om symptomer, hastigt hjernesvind, forløb, årsager og prognose for denne uhelbredelige neurologiske sygdom.
Creutzfeldt-Jakobs sygdom (udtales KROITS-felt YAH-kohb) eller CJD er en neurologisk sygdom. Den er degenerativ (den bliver værre med tiden), den kan ikke helbredes, og den fører altid til døden. CJD kaldes undertiden en menneskelig form for "kogalskab" (bovin spongiform encephalopati eller BSE). BSE er faktisk en årsag til en sjælden form for Creutzfeldt-Jakobs sygdom; de to er ikke den samme sygdom.
CJD skyldes et smittefremkaldendeagens kaldet en prion. Prioner er proteiner, der er foldet forkert. Prioner laver kopier af sig selv ved at ændre korrekt foldede proteiner til forkert foldede former. CJD får hjernevævet til at blive usundt meget hurtigt. Efterhånden som sygdommen ødelægger hjernen, opstår der huller i hjernen. Hjernens tekstur ændrer sig og bliver som en køkkensvamp.
Billedgalleri
6 Billeder




Typer af CJD og årsager
- Sporadisk CJD (sCJD): Den hyppigste form (ca. 85–90 % af tilfældene). Årsagen er ukendt, og sygdommen opstår uden kendt smitte eller arvelig mutation.
- Familær (arvelig) CJD: Forårsaget af mutationer i priongenet (PRNP). Denne form er autosomalt dominant og udgør omkring 10–15 % af tilfældene.
- Iatrogen CJD: Sjælden, sker via medicinske procedurer, f.eks. kontaminerede kirurgiske instrumenter, corneatransplantationer, dura mater-grafts eller tidligere brug af væksthormon udvundet fra humane hypofyser.
- Variant CJD (vCJD): En særskilt form forbundet med eksponering for BSE-smittet kødkød. vCJD rammer typisk yngre personer og har nogle kliniske forskelle fra sporadisk CJD.
Symptomer og forløb
CJD udvikler sig som regel hurtigt i forhold til andre demenssygdomme. Tidlige og hyppige symptomer omfatter:
- Hurtigt progredierende demens med nedsat hukommelse, koncentration og dømmekraft.
- Motoriske forstyrrelser såsom ukoordineret gang (ataxi) og stivhed.
- Myoklonier (hurtige muskelrykninger), som er karakteristiske og ofte ses tidligt.
- Syns- og taleforstyrrelser, forvirring og adfærdsændringer. Ved vCJD kan psykiatriske symptomer være fremtrædende i begyndelsen.
Forløbet er ofte hurtigt: mange patienter bliver alvorligt syge inden for få måneder og dør typisk inden for et år efter symptomdebut, skønt der er variation—nogle overlever længere tid.
Udredning og diagnose
Diagnosen bygger på kombination af klinisk billede og specialundersøgelser. Typiske diagnostiske værktøjer:
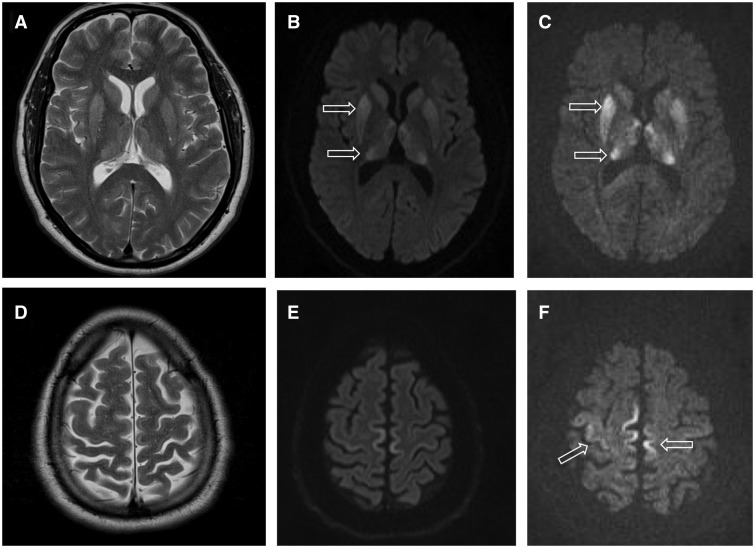
- MR-scanning (særligt diffusion-weighted imaging) der kan vise karakteristiske forandringer i cortex og basalganglier.
- EEG kan vise periodiske, skarpe bølger hos nogle patienter (ikke altid til stede).
- CSF-analyser (rygvæske): markører som 14-3-3-protein eller øget total tau kan støtte diagnosen, men er ikke fuldstændig specifikke.
- RT-QuIC (Real-Time Quaking-Induced Conversion): en nyere, meget sensitiv og specifik test, der påviser sygdomsfremkaldende prionaktivitet i cerebrospinalvæske eller næsesvælgprøver.
- Definitiv diagnose stilles ved neuropatologisk undersøgelse af hjernevæv (f.eks. efter død eller ved biopsi) med påvisning af spongiform forandring og ophobning af det abnorme prionprotein (PrPSc).
Behandling og pleje
Der findes ingen kurativ behandling for CJD. Behandlingen er derfor støttende og palliativ med fokus på symptomlindring og patientens komfort:
- Smertebehandling og pleje af almene behov.
- Behandling af myoklonier og kramper med lægemidler (fx benzodiazepiner eller antiepileptika) efter lægens vurdering.
- Ergoterapi, fysioterapi og støtte til ernæring og hygiejne så længe det er muligt.
- Palliativ indsats og samtaler med pårørende om prognose og plejevalg.
Forskning i anti-prion-lægemidler og immunterapier pågår, men indtil videre er der ingen dokumenteret effektiv behandling.
Smitsomhed og forebyggelse
Prioner er usædvanligt modstandsdygtige over for almindelig sterilisering. Derfor gælder strenge hygieneregler i sundhedsvæsenet ved håndtering af materiale fra patienter med kendt eller mistænkt CJD:
- Brug af engangsudstyr eller særlige decontaminationsprocedurer for instrumenter, der kan være kontamineret.
- Screening og overvågning af blod- og vævsdonationer samt fødevaresikkerhedsforanstaltninger for at undgå fødevarebaseret transmission (historisk relevant for BSE/vCJD).
- Informere sundhedspersonale og følge nationale og internationale retningslinjer for prævention.
Prognose og hyppighed
- Incidensen af sporadisk CJD er lav — omkring 1–2 tilfælde pr. million indbyggere årligt.
- Prognosen er ofte alvorlig med hurtig progression; dødelighed er høj og indtræder typisk inden for måneder til et år.
- vCJD har et noget anderledes forløb og rammer ofte yngre personer, men er meget sjælden.
Differentialdiagnoser
Andre sygdomme kan ligne CJD i starten, fx hurtigt progredierende Alzheimer, encephalitis, paraneoplastiske tilstande, metaboliske eller toksiske encefalopatier og visse andre neurodegenerative sygdomme. Derfor er grundig udredning nødvendig.
Råd til pårørende
- Søg løbende information fra behandlende læger og kommuniker åbent om plejemuligheder og prognose.
- Overvej tidlig kontakt til palliative teams for støtte til smertebehandling og efterlevelse af patientens ønsker.
- Husk at der findes nationale overvågningsprogrammer og rådgivning ved mistanke om prionsygdomme — sundhedspersonale kan henvise videre.
Bemærk: CJD er en sjælden, alvorlig sygdom. Hvis du eller en pårørende har symptomer, som vækker mistanke om hurtig neurologisk forværring, kontakt læge eller neurologisk afdeling for vurdering.
Typer og årsager til CJD
Typer af CJD omfatter:
- variant (vCJD):
Denne type CJD kan skyldes, at man spiser fødevarer, der indeholder prioner, f.eks. kød fra køer, der har BSE ("kogalskab"). Dette er dog en meget sjælden årsag til CJD.
- sporadisk (sCJD):
Dette er den mest almindelige type CJD. 85 % af tilfældene af CJD er sporadisk CJD. Ingen ved, hvad der forårsager sCJD; det ser ud til at ske tilfældigt.
- familiær (fCJD):
De fleste af de øvrige 15 % af CJD-tilfældene er familiær CJD. Det er en form for CJD, der forekommer i familier.
- iatrogen:
Denne form for CJD er normalt forårsaget af en medicinsk procedure, hvor en person får blod eller væv fra en person med CJD. En person kan f.eks. få iatrogen CJD, hvis han/hun får en blodtransfusion eller en hornhindetransplantation fra en person med CJD.
Tegn og symptomer
Det første symptom på CJD er demens, som forværres meget hurtigt. demens forårsager hukommelsestab, personlighedsændringer og hallucinationer.
Andre almindelige psykiske symptomer omfatter:
- Angst
- Depression
- Paranoia
- Obsessiv-kompulsive symptomer
- Psykose
Fysiske symptomer på CJD omfatter ofte:
- Problemer med at tale
- Rykkende bevægelser (myoklonus)
- Problemer med balancen (ataksi)
- Problemer med at gå
- Ryster eller er stiv
- Synsproblemer
- Synkebesvær, hvilket kan gøre det svært eller umuligt at spise
- Problemer med hoste, som kan forårsage lungebetændelse
- Bevægelser, som patienten ikke kan kontrollere (dyskinesi)
De fleste mennesker med CJD dør inden for seks måneder efter, at de første symptomer er opstået. Ofte dør de af lungebetændelse forårsaget af hosteproblemer. Omkring 15 % af patienterne overlever i to år eller mere. Nogle patienter har levet 4-5 år med mest mentale symptomer, indtil sygdommen bliver værre og forårsager flere fysiske symptomer. Når dette sker, dør folk normalt inden for et år.
Symptomerne på CJD skyldes, at flere og flere af hjernens nerveceller dør. Når forskere ser på hjernevæv fra en CJD-patient under et mikroskop, kan de se mange små huller, hvor hele områder af nerveceller er døde.
Diagnoser
Læger kan have mistanke om CJD, når en person har visse symptomer. For eksempel forværres demens normalt langsomt. Demens, der forværres meget hurtigt, er usædvanligt. Sammen med symptomer som rykagtige bevægelser kan disse symptomer pege på mulig CJD.
Derefter kan man foretage prøver for at vise, om personen har CJD. Disse prøver omfatter:
- Elektroencefalografi (EEG): Denne test viser den elektriske aktivitet i hjernen. En læge vil ofte kunne se ændringer i EEG'et, som er almindelige hos personer med CJD. Den type ændringer, der vises på EEG'et, afhænger af, hvilken type CJD patienten har, og hvor langt sygdommen er fremme.
- Lumbalpunktur (rygmarvsprøve): Denne test gør det muligt at undersøge cerebrospinalvæske (den væske, der omgiver hjernen og rygmarven) for at finde et bestemt protein ("14-3-3-protein").
- MRI af hjernen: En test, der bruger en meget stærk magnet til at tage billeder af hjernen
- Biopsi: Ved en biopsi bruger en kirurg en nål til at tage et lille stykke væv fra kroppen, så lægerne kan se det i et mikroskop. vCJD kan diagnosticeres ved hjælp af en biopsi af mandlerne. For alle de andre typer CJD er en biopsi af hjernen den eneste måde, hvorpå man med sikkerhed kan fastslå, om en person har CJD. Da en biopsi af hjernen imidlertid kan forårsage hjerneskade, foretages en hjernebiopsi normalt ikke, hvis andre undersøgelser allerede har vist, at en person sandsynligvis har CJD.

Behandling
I 2016 findes der ingen behandling, der kan helbrede CJD eller endda bremse virkningerne. Der foretages mange forsøg for at finde behandlinger.
I dag er den eneste behandling af CJD medicin, som behandler sygdommens symptomer og hjælper patienterne med at få det bedre. Patienter med krampeanfald kan f.eks. få krampestillende medicin. Benzodiazepiner kan få muskelrykningerne til at blive mindre hyppige.
Patienterne kan også vælge at få medicinske procedurer for at hjælpe med dårlige symptomer. CJD kan f.eks. forårsage så store problemer med at synke, at en person ikke kan spise. Nogle mennesker med CJD vælger at få lagt en sonde til fodring, når de ikke længere kan spise. Det er et rør, der går ind i maven, så der kan gives en særlig væske direkte ind i maven for at give personen næring.
Relaterede sider
- Prion
- Prionsygdom
- Terminal sygdom
Spørgsmål og svar
Spørgsmål: Hvad er Creutzfeldt-Jakobs sygdom?
A: Creutzfeldt-Jakobs sygdom (CJD) er en neurologisk sygdom, der er degenerativ, uhelbredelig og altid dødelig.
Sp: Er der en kur mod CJD?
A: Nej, der findes ingen kur mod CJD.
Spørgsmål: Hvorfor omtales CJD nogle gange som en menneskelig form for "kogalskab"?
A: CJD kaldes nogle gange en menneskelig form for "kogalskab", fordi bovin spongiform encephalopati (BSE), som er årsag til en sjælden form for CJD, almindeligvis kaldes "kogalskab".
Sp: Hvad er årsagen til CJD?
A: CJD skyldes et smittefremkaldende agens kaldet et prion, som er et protein, der er foldet forkert og kan lave kopier af sig selv ved at ændre korrekt foldede proteiner til forkert foldede proteiner.
Sp: Hvad sker der med hjernevævet ved CJD?
Svar: CJD medfører, at hjernevævet meget hurtigt bliver usundt, hvilket resulterer i udvikling af huller i hjernen og en ændring af hjernens tekstur, så den ligner en køkkensvamp.
Spørgsmål: Er BSE den samme sygdom som CJD?
A: Nej, BSE er ikke den samme sygdom som CJD; det er faktisk en årsag til en sjælden type CJD.
Spørgsmål: Hvordan forårsager prioner CJD?
Svar: Prioner forårsager CJD ved at folde sig forkert og lave kopier af sig selv på bekostning af korrekt foldede proteiner i hjernen. Dette resulterer i ødelæggelse af sundt hjernevæv og udvikling af de huller, der er karakteristiske for sygdommen.
Relaterede artikler
Forfatter
AlegsaOnline.com Creutzfeldt-Jakobs sygdom (CJD) – dødelig prionsygdom: symptomer og forløb Leandro Alegsa
URL: https://da.alegsaonline.com/art/24152
Kilder
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"