Pulmonal arteriel hypertension (PAH): Årsager, symptomer og behandling
Lær om pulmonal arteriel hypertension (PAH): årsager, symptomer, diagnose og moderne behandlinger. Få viden om tidlige tegn, risikofaktorer og forbedring af livskvalitet.
Pulmonal hypertension (PH) er en sygdom, hvor blodtrykket i blodkarrene i lungerne er forhøjet. Den øgede modstand i lungekredsløbet gør det sværere for hjertet at pumpe blodet videre til lungerne og resten af kroppen. Når belastningen bliver for stor, kan hjertet svækkes, og der kan udvikles hjertesvigt. I nogle tilfælde kan tilstanden blive så alvorlig, at en lungetransplantation eller en hjerte-lunge-transplantation bliver nødvendig for at redde livet.
Billedgalleri
9 Billeder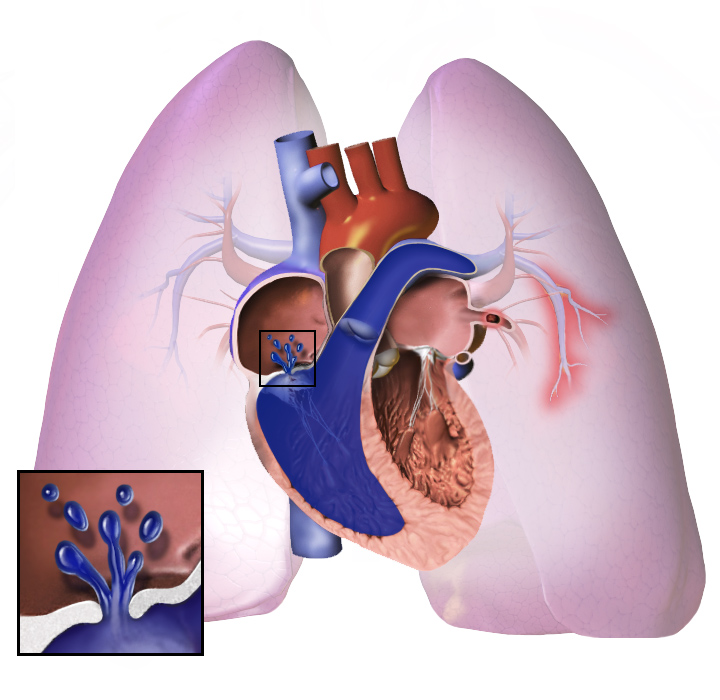






Hvad er forskellen på PH og PAH?
Pulmonal arteriel hypertension (PAH) er en undergruppe af pulmonal hypertension. Begrebet PH dækker over flere forskellige årsager til forhøjet tryk i lungekarrene, mens PAH beskriver den form, hvor sygdommen primært sidder i arterierne i lungerne. Mange taler kort og kalder tilstanden PAH, PH eller PHA, men det er vigtigt at få klarlagt, hvilken type der er tale om, fordi behandling og prognose kan være forskellig.
Årsager
PH/PAH kan opstå af mange forskellige grunde. Almindelige årsager og risikofaktorer omfatter:
- Idiopatisk (ukendt årsag) eller arvelig PAH
- Kollagenoser og bindevævssygdomme (fx sklerodermi)
- Medfødte hjertesygdomme, der påvirker blodgennemstrømningen
- Leversygdom med portal hypertension
- Infektioner som HIV eller schistosomiasis
- Visse medicin og stoffer (fx tidligere vægt-tab præparater som fenfluramin)
- Langvarig lav iltmætning (fx ved svær lungesygdom)
- Blodprop i lungerne, som giver kronisk tromboembolisk pulmonal hypertension (CTEPH)
Symptomer
Symptomerne kommer ofte gradvist og kan være diffuse tidligt i forløbet. De mest almindelige er:
- Åndenød ved anstrengelse eller i hvile (trække vejret svært)
- Unormal træthed og nedsat fysisk formåen
- Svimmelhed eller besvimelser (besvimer let)
- Brystsmerter eller trykken for brystet
- Hævelse i benene (ødem), oppustet mave ved væskeophobning
Symptomerne bliver ofte værre ved fysisk aktivitet. Nogle patienter har behov for ekstra ilt.
Undersøgelser og diagnose
Hvis mistanke om PH/PAH opstår, vil lægen ofte starte med:
- Blodprøver og almindelig klinisk undersøgelse
- Røntgen eller CT-scanning af thorax
- Ekkokardiografi (ultralyd af hjertet) som screeningsundersøgelse
- Hjertekateterisation af højre side (right heart catheterization) — guldstandard for at måle trykket i lungearterierne og stille en sikker diagnose
- Yderligere tests for at finde årsagen: lungefunktionsmåling, V/Q-scintigrafi for at udelukke CTEPH, HIV-test, leverprøver m.m.
Behandling
Behandlingen afhænger af årsagen og sygdommens sværhedsgrad. Målet er at sænke blodtrykket i lungekredsløbet, forbedre symptomer og mindske risikoen for hjertesvigt. Typiske tiltag og behandlinger:
- Generelle råd: undgå overanstrengelse, få vaccination mod influenza og pneumokokker, behandle øvrige sygdomme
- Oxygenbehandling ved lav iltmætning
- Diuretika (vanddrivende medicin) ved væskeophobning
- Antikoagulation (blodfortyndende) kan anbefales ved visse typer, især ved CTEPH
- Calciumkanalblokkere kan hjælpe hos få patienter, der reagerer positivt ved specielle tests
- PAH-specifikke lægemidler:
- Endotelinreceptorantagonister (fx bosentan, ambrisentan)
- Fosfodiesterase-5-hæmmere (fx sildenafil, tadalafil)
- Riociguat (soluble guanylate cyclase-stimulator)
- Prostacyclin-analoger (fx epoprostenol, treprostinil, iloprost) og prostacyclinreceptoragonist (selexipag)
- Ved kronisk tromboembolisk pulmonal hypertension (CTEPH) kan kirurgisk fjernelse af blodpropper (pulmonal endarterektomi) være helbredende, og riociguat kan bruges, hvis operation ikke er mulig
- I fremskredne tilfælde kan lungetransplantation eller hjerte-lunge-transplantation være nødvendig
- Nogle patienter kan få palliativ procedure som atrial septostomi i særlige tilfælde for at aflaste højre hjerte
Opfølgning og prognose
PAH/PH er en kronisk sygdom, der kræver regelmæssig opfølgning på et specialcenter. Prognosen varierer meget afhængig af årsag, hvor tidligt sygdommen opdages, og hvor hurtigt man får relevant behandling. Moderne målrettet behandling har forbedret livskvalitet og overlevelse markant, men sygdommen kan stadig være livstruende, især uden behandling.
Særlige forhold
- Graviditet er risikofyldt ved PAH og frarådes ofte på grund af stor risiko for mor og foster
- Hvis du har kendte risikofaktorer eller symptomer, er det vigtigt at søge læge tidligt
- Undgå medikamenter og stoffer, der er kendt for at øge risikoen for PAH
Hvornår skal du søge læge?
Søg lægehjælp ved vedvarende eller forværrede symptomer som åndenød, besvimelser, brystsmerter eller pludselig vægtøgning pga. væskeophobning. Tidlig udredning kan føre til tidligere behandling og bedre resultat.
Hvis du vil vide mere, kan din praktiserende læge henvise dig til et specialiseret pulmonal hypertension-center, hvor der er erfaring med udredning og moderne behandling. Pulmonal hypertension er alvorligt, men korrekt diagnose og behandling kan give mange patienter et bedre og længere liv.
Tegn og symptomer
Personer med pulmonal hypertension har svært ved at trække vejret. De bliver også let trætte. Nogle af dem besvimer også let. De kan have smerter i brystet. Nogle patienter har hævelse af fødder og ankler. Disse symptomer bliver værre under motion eller hårdt arbejde.
Da mange sygdomme kan gøre det svært at trække vejret, skal lægen lære om patientens baggrund. Dette hjælper lægen med at behandle patienten, også selv om patienten har en anden sygdom. Lægen foretager også flere prøver. Pulmonal hypertension får hjertet til at lyde anderledes. En af testene er at måle blodtrykket inde i lungearterien, det blodkar, der går fra hjertet til lungerne.
For at fastslå årsagen vil lægen som regel foretage en grundig anamnese for at finde frem til årsagen. Der tages en detaljeret familiehistorie for at afgøre, om sygdommen kan være familiær. En historie med udsættelse for kokain, methamfetamin, alkohol, der fører til skrumpelever, og rygning, der fører til emfysem, anses for at være af betydning. Der foretages en fysisk undersøgelse for at finde typiske tegn på pulmonal hypertension, herunder en høj P2-lyd (lukkelyd fra pulmonalklappen), (para)sternal hævning, udspilning af jugularvenen, pedalødem, ascites, hepatojugulær refluks, clubbing osv.
Hvad går galt med kroppen
Ved pulmonal hypertension bliver blodkarrene i lungerne for snævre. Blodtrykket i lungerne bliver højt. Hjertet arbejder meget hårdt for at pumpe blodet gennem de snævre blodkar. Senere bliver blodkarrene i lungerne hårde og tykke. Hjertet skal arbejde hårdere.
Hjertet kan arbejde så hårdt, at det bliver sygt. Dette kaldes hjertesvigt. Det syge hjerte kan ikke pumpe blodet godt. Der kommer mindre blod til lungerne, så blodet får mindre ilt. Det gør det svært at trække vejret. Det bliver værre, når man træner eller arbejder hårdt.
Årsager
Den mest almindelige årsag til pulmonal hypertension er venstre hjertesvigt. Dette forårsager pulmonalvenøs hypertension. Dette fører til lungeødem eller væskeophobning i lungerne.
Mange sygdomme kan forårsage pulmonal arteriel hypertension (PAH).
- Lungesygdomme, der medfører, at blodet får mindre ilt, f.eks:
· kronisk obstruktiv lungesygdom eller COPD
· interstitiel lungesygdom
· Pickwickian syndrom
- problemer med immunsystemet, såsom:
· AIDS
· sklerodermi
· andre autoimmune sygdomme
- leverproblemer
· skrumpelever
· portal hypertension
- andre årsager
· søvnapnø
· at tage piller for at tabe sig, såsom Fen-Phen, Aminorex, fenfluramin (Pondimin) og phentermin
· seglcellesygdom,
· medfødt hjertesygdom
· sygdomme i skjoldbruskkirtlen,
· tage stoffer som kokain
· muligvis Human herpesvirus 8
Når en person har pulmonal hypertension uden nogen anden årsag, kaldes det idiopatisk pulmonal arteriel hypertension eller IPAH.
Når der findes en familiehistorie, kaldes sygdommen for familiær pulmonal arteriel hypertension (FPAH). IPAH og FPAH anses nu for at være genetiske sygdomme, der er knyttet til mutationer i BMPR2-genet, som koder for en receptor for knoglemorfogenetiske proteiner, samt i 5-HT(2B)-genet, som koder for en serotoninreceptor.
Inden for medicin er pulmonal hypertension (PH) en stigning i blodtrykket i lungearterien eller lungekarrene, hvilket fører til åndenød, svimmelhed, besvimelse og andre symptomer, som alle forværres ved anstrengelse. Afhængigt af årsagen kan pulmonal hypertension være en alvorlig sygdom med en markant nedsat træningstolerance og højresidig hjertesvigt. Den blev først identificeret af Dr. Ernst von Romberg i 1891. Den kan være en af fem forskellige typer: arteriel, venøs, hypoxisk, tromboembolisk eller forskelligartet.
Selv om udtrykkene primær pulmonal hypertension (dvs. af ukendt årsag) og sekundær pulmonal hypertension (dvs. som følge af en anden medicinsk tilstand) stadig findes i materiale, der formidles til patienter og offentligheden, er disse udtryk stort set blevet opgivet i den medicinske litteratur. Denne ændring er sket, fordi den ældre dikotomiske klassifikation ikke afspejlede patofysiologien eller resultatet. Den førte til fejlagtige terapeutiske beslutninger, dvs. at man kun behandlede "primær" pulmonal hypertension. Dette førte til gengæld til terapeutisk nihilisme for mange patienter, der blev betegnet som "sekundær" pulmonal hypertension, og det kunne have bidraget til deres død. Udtrykket "primær pulmonal hypertension" er nu blevet erstattet med "idiopatisk pulmonal arteriel hypertension". Begreberne "primær" og "sekundær" pulmonal hypertension bør ikke længere anvendes. Yderligere oplysninger findes i afsnittet om klassifikation nedenfor.
Årsager
Den mest almindelige årsag til pulmonal hypertension er venstre hjertesvigt, der fører til pulmonalvenøs hypertension. Dette kan skyldes systolisk eller diastolisk fejlfunktion i venstre hjertekammer eller ventildysfunktion som f.eks. mitralinsufficiens eller mitralstenose. Den viser sig normalt som lungeødem.
Almindelige årsager til pulmonal arteriel hypertension (PAH) omfatter HIV, sklerodermi og andre autoimmune sygdomme, skrumpelever og portal hypertension, seglcellesygdom, medfødte hjertesygdomme, skjoldbruskkirtelsygdomme og andre. Brug af vægttabspiller som Fen-Phen, Aminorex, fenfluramin (Pondimin) og phentermin har tidligere ført til udvikling af PAH.
Patogenese
Uanset den oprindelige årsag indebærer pulmonal hypertension en stramning af blodkarrene i forbindelse med og i lungerne. Dette gør det sværere for hjertet at pumpe blodet gennem lungerne, ligesom det er sværere at få vand til at strømme gennem et smalt rør end gennem et bredt rør. Med tiden bliver de berørte blodkar både stivere og tykkere, hvilket øger blodtrykket i lungerne yderligere og forringer blodgennemstrømningen. Desuden medfører den øgede arbejdsbelastning af hjertet en fortykkelse og forstørrelse af højre hjertekammer, hvilket gør hjertet mindre i stand til at pumpe blod gennem lungerne, hvilket forårsager højre hjertesvigt. Da blodet, der strømmer gennem lungerne, mindskes, modtager venstre side af hjertet mindre blod. Dette blod kan også transportere mindre ilt end normalt. Derfor bliver det sværere og sværere for venstre side af hjertet at pumpe for at levere tilstrækkelig ilt til resten af kroppen, især under fysisk aktivitet.
Diagnoser
Da pulmonal hypertension kan være af 5 hovedtyper, skal der udføres en række test for at skelne mellem pulmonal arteriel hypertension og venøs, hypoxisk, thomboembolisk eller diverse varianter.
Der foretages en fysisk undersøgelse for at se efter typiske tegn på pulmonal hypertension. Disse omfatter ændrede hjertelyd, f.eks. et bredt delt S2 eller anden hjertelyd, et højt P2 eller pulmonalklaplukkelyd (en del af anden hjertelyd), (para)sternal hævning, mulig S3 eller tredje hjertelyd og pulmonal regurgitation. Andre tegn er bl.a. jugular venus distension (udvidelse af jugularvenerne), perifert ødem (hævelse af ankler og fødder), ascites (abdominal hævelse på grund af væskeophobning), hepatojugulær refluks og klynk.
Der er behov for yderligere procedurer for at bekræfte tilstedeværelsen af pulmonal hypertension og udelukke andre mulige diagnoser. Disse omfatter generelt lungefunktionstest, blodprøver, elektrokardiografi (EKG), måling af arterielle blodgasser, røntgenbilleder af brystkassen (efterfulgt af CT-scanning med høj opløsning, hvis der er mistanke om interstitiel lungesygdom) og ventilation-perfusion eller V/Q-scanning for at udelukke kronisk tromboembolisk pulmonal hypertension. Biopsi af lungerne er normalt ikke indiceret, medmindre man mener, at den pulmonale hypertension skyldes en underliggende interstitiel lungesygdom. Men lungebiopsier er behæftet med risiko for blødning på grund af det høje intrapulmonale blodtryk. Klinisk forbedring måles ofte ved en "seksminute walk test", dvs. den afstand, som en patient kan gå på seks minutter. Stabilitet og forbedring i denne måling korrelerer med bedre overlevelse.
Selv om pulmonalarterietrykket kan estimeres på grundlag af ekkokardiografi, giver trykprøvetagning med et Swan-Ganz-kateter den mest sikre måling. PAOP og PVR kan ikke måles direkte med ekkokardiografi. Derfor kræver diagnosen PAH en hjertekateterundersøgelse. Et Swan-Ganz-kateter kan også måle hjertevolumen, som er langt vigtigere for måling af sygdommens sværhedsgrad end det pulmonale arterietryk.
Det normale pulmonale arterietryk hos en person, der lever på havniveau, har en middelværdi på 12-16 mm Hg (1600-2100 Pa). Der er tale om decideret pulmonal hypertension, når middeltrykket i hvile overstiger 25 mm Hg (3300 Pa). Hvis pulmonalarteriemedietrykket stiger til over 30 mm Hg (4000 Pa) ved anstrengelse, betragtes dette også som pulmonal hypertension.
Diagnosen PAH kræver tilstedeværelse af pulmonal hypertension sammen med to andre tilstande. Pulmonalarterie-okklusionstrykket (PAOP eller PCWP) skal være mindre end 15 mm Hg (2000 Pa) og pulmonalvaskulær modstand (PVR) skal være større end 3 Wood-enheder (240 dyn-s-cm−5 eller 2,4 mN-s-cm−5 ).
Klassifikation
Nuværende klassificering
I 2003 blev det tredje verdenssymposium om pulmonal arteriel hypertension afholdt i Venedig for at ændre klassifikationen på baggrund af den nye forståelse af sygdomsmekanismerne. Det reviderede system, der blev udviklet af denne gruppe, udgør den nuværende ramme for forståelsen af pulmonal hypertension.
Systemet indeholder flere forbedringer i forhold til det tidligere Evian-klassifikationssystem fra 1998. Beskrivelserne af risikofaktorer er blevet opdateret, og klassifikationen af medfødte systemiske til pulmonale shunts er blevet revideret. Der blev anbefalet en ny klassifikation af genetiske faktorer i PH, men den blev ikke gennemført, fordi de tilgængelige data blev anset for at være utilstrækkelige.
Det reviderede klassifikationssystem fra Venedig 2003 kan sammenfattes som følger:
- WHO-gruppe I - Pulmonal arteriel hypertension (PAH)
- WHO-gruppe II - Pulmonal hypertension i forbindelse med venstre hjertesygdom
- WHO-gruppe III - Pulmonal hypertension i forbindelse med lungesygdomme og/eller hypoxæmi
- WHO-gruppe IV - Pulmonal hypertension som følge af kronisk trombotisk og/eller embolisk sygdom
- WHO-gruppe V - Diverse
Tidligere terminologi
Tidligere blev begreberne primær og sekundær pulmonal hypertension (PPH og SPH) anvendt til at klassificere sygdommen. Dette førte til den antagelse, at kun den primære sygdom skulle behandles, og at den sekundære variant skulle ignoreres til fordel for behandling af kun den underliggende sygdom. I virkeligheden kan alle former for pulmonal arteriel hypertension behandles. Desværre er dette klassifikationssystem stadig i mange lægers bevidsthed, og det fører sandsynligvis til, at mange patienter nægtes behandling. Denne nihilistiske tilgang til pulmonal arteriel hypertension kan også bidrage til underdiagnosticering. Det anslås, at der er omkring 100 000 patienter med PAH i USA, men kun 15-20 000 er blevet diagnosticeret. Mange andre er blevet fejldiagnosticeret som KOL, astma eller kongestiv hjertesvigt.
Udtrykket primær pulmonal hypertension (PPH) er nu blevet erstattet med idiopatisk pulmonal arteriel hypertension (IPAH) i en stor del af den medicinske litteratur. Nogle læger fortsætter dog med at bruge den ældre klassifikation uhensigtsmæssigt.
Epidemiologi
IPAH er en sjælden sygdom med en incidens på ca. 2-3 pr. million om året og en prævalens på ca. 15 pr. million. Kvinder har næsten tre gange så stor sandsynlighed for at få IPAH som mænd.
Andre former for PAH er langt mere almindelige. Forekomsten er anslået til 6-60 % af alle patienter med sklerodermi, op til 21 % med reumatoid arthritis, 4-14 % med systemisk lupus erythematosus, 2-5 % med portal hypertension, 0,5 % med HIV og 20-40 % med seglcellesygdom.
Diætpiller som Fen-Phen havde en årlig forekomst på 25-50 pr. million pr. år.
Behandling
Behandlingen bestemmes af, om PH er arteriel, venøs, hypoxisk, tromboembolisk eller forskelligartet. Da pulmonalvenøs hypertension er synonymt med kongestiv hjertesvigt, er behandlingen at optimere venstre ventrikelfunktionen ved hjælp af diuretika, betablokkere, ACE-hæmmere osv. eller at reparere/udskifte mitralklappen eller aortaklappen.
Ved PAH betragtes livsstilsændringer, digoxin, diuretika, orale antikoagulantia og iltbehandling som konventionel behandling, men det er aldrig blevet bevist, at de har været gavnlige på en randomiseret, prospektiv måde.
Højt doserede calciumkanalblokkere er kun nyttige hos 5 % af IPAH-patienter, som er vasoreaktive ved Swan-Ganz-kateter. Desværre er kalciumkanalblokkere i vid udstrækning blevet misbrugt, idet de er blevet ordineret til mange patienter med ikke-vasoreaktiv PAH, hvilket har ført til overdreven morbiditet og mortalitet.
Vasoaktive stoffer
Tre hovedveje er involveret i unormal proliferation og sammentrækning af glatmuskelcellerne i lungearterien hos patienter med pulmonal arteriel hypertension. Disse veje svarer til vigtige terapeutiske mål i denne tilstand og spiller en rolle ved bestemmelsen af, hvilken af tre klasser af lægemidler - endothelinreceptorantagonister, fosfodiesterase type 5-hæmmere og prostacyclinderivater - der skal anvendes.
Prostacyclin (prostaglandin I2 ) anses almindeligvis for at være den mest effektive behandling af PAH. Epoprostenol (syntetisk prostacyclin, markedsført som Flolan®) gives via kontinuerlig infusion, som kræver et semi-permanent centralt venekateter. Dette administrationssystem kan forårsage sepsis og trombose. Flolan® er ustabilt og skal derfor opbevares på is under administrationen. Da det har en halveringstid på 3-5 minutter, skal infusionen være kontinuerlig (døgnet rundt), og en afbrydelse kan være fatal. Der er derfor blevet udviklet andre prostanoider. Treprostinil (Remodulin®) kan gives intravenøst eller subkutant, men den subkutane form kan være meget smertefuld. Iloprost (Ilomedin®) anvendes også i Europa intravenøst og har en længere halveringstid. Iloprost (markedsført som Ventavis®) er den eneste inhalerede form af prostacyklin, der er godkendt til brug i USA og Europa. Denne indgiftsform har den fordel, at den har en selektiv deponering i lungerne med færre systemiske bivirkninger.
Den dobbelte (ETA og ETB ) endothelinreceptorantagonist bosentan (markedsført som Tracleer®) blev godkendt i 2001. To selektive endothelinreceptorantagonister (kun ETA ) er i den sidste fase af godkendelsesproceduren: sitaxsentan og ambrisentan. Sildenafil, en selektiv hæmmer af cGMP-specifik fosfodiesterase type 5 (PDE5), blev godkendt til behandling af PAH i 2005. Det markedsføres til PAH under navnet Revatio®. Tadalafil (der i øjeblikket markedsføres som Cialis® til behandling af erektil dysfunktion) er i øjeblikket i fase III-forsøg. Vasoaktivt intestinal peptid ved inhalation bør indgå i kliniske forsøg til behandling af PAH i 2007. PRX-08066 er en serotoninantagonist, der i øjeblikket er under udvikling til behandling af hypoxisk pulmonal hypertension.
Kirurgisk
Atrial septostomi er et kirurgisk indgreb, der skaber en forbindelse mellem højre og venstre forkammer. Det mindsker trykket på højre side af hjertet, men på bekostning af et lavere iltniveau i blodet (hypoxi). Den udføres bedst på erfarne centre. Lungetransplantation helbreder pulmonal arteriel hypertension, men efterlader patienten med de komplikationer, der følger med transplantationen, og en overlevelsestid på ca. 5 år.
Pulmonal tromboendarterektomi (PTE) er et kirurgisk indgreb, der anvendes ved kronisk tromboembolisk pulmonal hypertension. Det er en kirurgisk fjernelse af en organiseret trombus (blodprop) sammen med foringen af lungearterien; det er et stort og meget vanskeligt indgreb, som i øjeblikket udføres på nogle få udvalgte centre. Caseserier viser bemærkelsesværdig succes hos de fleste patienter.
Behandling af hypoxiske og forskellige former for pulmonal hypertension er ikke blevet fastlagt. Der er dog i øjeblikket undersøgelser af flere midler i gang, som omfatter patienter. Mange læger vil behandle disse sygdomme med den samme medicin som ved PAH, indtil der kommer bedre muligheder til rådighed.
Prognose
NIH IPAH-registeret fra 1980'erne viste en ubehandlet medianoverlevelse på 2-3 år fra diagnosetidspunktet, hvor dødsårsagen normalt var højre ventrikelsvigt (cor pulmonale). Selv om dette tal er almindeligt citeret, er det sandsynligvis irrelevant i dag. Resultaterne har ændret sig dramatisk i løbet af de sidste to årtier. Dette kan skyldes nyere lægemiddelbehandling, bedre generel pleje og tidligere diagnosticering (lead time bias). En nylig undersøgelse af resultaterne af de patienter, der havde påbegyndt behandling med bosentan (Tracleer®), viste, at 86 % af patienterne var i live efter 3 år. Da der nu er flere midler til rådighed, anvendes kombinationsbehandling i stigende grad. Disse midlers indvirkning på overlevelsen kendes ikke, da mange af dem først er blevet udviklet for nylig. Det ville ikke være urimeligt at forvente, at medianoverlevelsen i den nærmeste fremtid vil stige til over 10 år.
Spørgsmål og svar
Q: Hvad er pulmonal hypertension eller PH?
A: Pulmonal hypertension eller PH er en tilstand, hvor der er højt blodtryk i lungerne.
Q: Hvad er symptomerne på pulmonal hypertension?
A: Symptomerne på pulmonal hypertension omfatter åndedrætsbesvær, svimmelhed, træthed og besvimelse.
Q: Hvorfor har nogle mennesker med pulmonal hypertension brug for ekstra ilt?
A: Nogle mennesker med pulmonal hypertension har brug for ekstra ilt, fordi tilstanden gør det svært for dem at trække vejret.
Q: Hvornår bliver symptomerne på pulmonal hypertension værre?
A: Symptomerne på pulmonal hypertension bliver værre, når man træner eller arbejder hårdt.
Q: Hvorfor er pulmonal hypertension en alvorlig tilstand?
A: Pulmonal hypertension er en alvorlig tilstand, fordi den gør det sværere for hjertet at pumpe blod og kan være dødelig.
Q: Hvad er det fulde navn for pulmonal hypertension?
A: Det fulde navn for pulmonal hypertension er pulmonal arteriel hypertension, selv om de fleste kalder det pah, ph eller pha.
Q: Hvad kan nogle meget syge mennesker med pulmonal hypertension have brug for for at leve?
A: Nogle meget syge mennesker med pulmonal hypertension kan have brug for en lungetransplantation eller en hjerte-lunge-transplantation for at leve.
Relaterede artikler
Forfatter
AlegsaOnline.com Pulmonal arteriel hypertension (PAH): Årsager, symptomer og behandling Leandro Alegsa
URL: https://da.alegsaonline.com/art/80022
Kilder
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171