Progeroide syndromer (progeria): årsager, symptomer og genetik
Få overblik over progeroide syndromer (progeria): årsager, symptomer, genetik, mutationer i lamin A/C og DNA-reparation, prognose og aktuel forskning.
Progeroide syndromer (PS) beskriver en række genetiske sygdomme, hvor den ramte person synes at blive hurtigere ældre. Alle disse sygdomme er monogenetiske, hvilket betyder, at de skyldes mutationer i et enkelt gen. De fleste kendte PS-mutationer fører enten til defekter i DNA-reparationsmekanismen eller defekter i et protein, der kaldes lamin A/C.
Progeroid betyder "som ligner alderdom". Denne betegnelse kan anvendes på mange forskellige sygdomme. Alzheimerssygdom og Parkinsons sygdom påvirker ofte kun ét væv. I de fleste tilfælde bruges udtrykket progeroid syndrom om tilstande, hvor de berørte personer viser flere træk, man normalt forbinder med aldring (fx hårtab, tynd hud, knogleskørhed), men ikke nødvendigvis alle aldringstegn, og hvor flere vævstyper er påvirkede.
Personer med PS-relaterede lidelser har ofte en kortere levetid. De mest studerede progeroide syndromer er Werner syndromet (WS) og Hutchinson-GilfordProgeria syndromet (HGPS), fordi de ligner naturlig aldring. På grund af deres karakteristika med accelereret aldring (senescens) er progeroide syndromer blevet flittigt studeret inden for aldringsforskning, regeneration, stamceller og kræft.
Billedgalleri
3 Billeder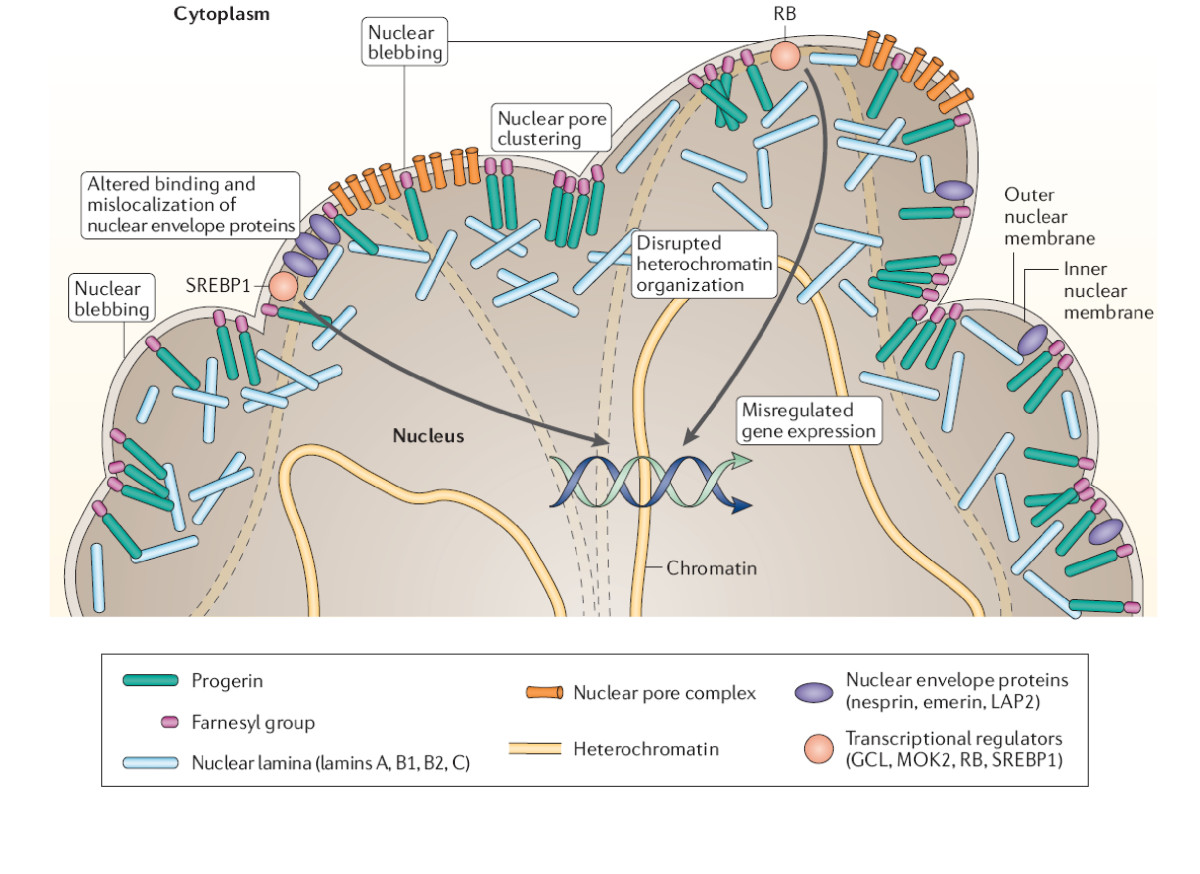

Årsager og genetik
Progeroide syndromer har forskellige underliggende årsager, men to hovedmekanismer går igen:
- Defekter i DNA-reparation: Mutationer i gener, der er ansvarlige for at reparere skader i DNA, fører til genomisk ustabilitet, øget celledød og funktionssvigt i væv. Eksempler inkluderer gener, der er involveret i nukleotid-excision-reparation (som ved Cockayne-syndrom og i nogle aspekter hos patienter med xeroderma pigmentosum), samt RECQ‑helikaser som WRN (Werner) og BLM (Bloom).
- Defekter i lamin A/C og nuklear struktur: Lamin A/C er en del af cellekernen's indre skelet. Mutationer i LMNA eller i enzymer, der processerer prelamin A (fx ZMPSTE24), fører til dannelse af en forkortet, toksisk form af lamin A kaldet progerin. Dette ses karakteristisk ved Hutchinson-GilfordProgeria syndromet (HGPS), hvor nukleære deformationer, ændret genudtryk og cellestress medfører accelereret vævsdegeneration.
Arvegangen varierer mellem syndromerne:
- HGPS skyldes typisk en de novo (ny) punktmutation i LMNA og opfører sig som autosomal dominant, men næsten alle tilfælde er ikke-arvelige, idet mutationerne opstår spontant.
- Werner, Bloom, Cockayne og mange andre progeroide syndromer arves ofte autosomalt recessivt, hvilket betyder, at begge kopier af genet skal være muterede for at sygdommen udvikles.
Typiske syndromer og deres kendetegn
- Hutchinson‑Gilford Progeria (HGPS) — meget sjælden; børn ser normale ud ved fødslen, men udvikler tidlig væksthæmning, tab af underhudsfedt, skaldethed, karakteristisk ansigtsudtryk, stive led og fremskreden åreforkalkning. Dødsårsagen er ofte hjerte-kar sygdom (myokardieinfarkt eller slagtilfælde) i barne- eller teenageårene.
- Werner syndrom — debuterer i ung voksentilstand (20’erne–30’erne). Karakteristika: grå stær, hårgråning, hudforandringer, osteoporose, sene sårhelingsproblemer, øget risiko for type 2‑diabetes og visse kræftformer. Livsforventningen er reduceret (ofte midt i 40’erne–50’erne) hovedsageligt pga. kræft og hjerte-kar sygdom.
- Bloom syndrom og Cockayne syndrom — begge associeret med væksthæmning og øget følsomhed over for DNA‑skade, men med forskellige kliniske mønstre (fx fotosensitivitet og neurologisk påvirkning ved Cockayne).
- Xeroderma pigmentosum (XP) — primært kendt for ekstrem følsomhed over for UV‑lys og kraftigt øget risiko for hudkræft; nogle patienter viser også progeroide træk i huden.
Symptomer og kliniske træk
Selvom symptomer varierer efter syndrom, ses ofte:
- Væksthæmning og lille kropsbygning
- Tynd eller skinnende hud, tab af underhudsfedt
- Hårtab (alopecia), tidlig grånelse
- Knogleskørhed (osteoporose), ledstivhed og kontrakturer
- Katarakt (grå stær), syns- og tandproblemer
- Øget tendens til sår, sårhelingsproblemer og nogle gange kræft
- Hjerte-kar sygdom, især hos HGPS, som er en ledende dødsårsag
Diagnose
Diagnosen bygger på en kombination af klinisk vurdering og genetisk testning. Typiske skridt:
- Grundig klinisk undersøgelse med dokumentation af vækst, hud, hår, øjne, knogler og neurologisk funktion.
- Molekylærgenetisk analyse af mistænkte gener (fx LMNA, WRN, BLM, ERCC6/ERCC8 m.fl.) for at bekræfte diagnosen.
- Supplerende undersøgelser som hjerte-/kar‑screening (ekko, arteriosklerosevurdering), DXA-scanning for knogletæthed, øjenundersøgelse mv.
Behandling og pleje
Der findes sjældent helbredende behandlinger for progeroide syndromer; pleje er ofte symptomatisk og tværfaglig:
- Kardiovaskulær overvågning og behandling er afgørende, især ved HGPS (blodtryk, kolesterol, tromboseforebyggelse ved behov).
- Sårbehandling, fodpleje og ortopædisk opfølgning ved problemer med sår og led.
- Endokrinologisk behandling ved diabetes eller andre stofskiftesygdomme (hyppigt ved Werner).
- Kirurgiske indgreb efter behov (fx kataraktoperation ved tidlig grå stær).
- Specifikke lægemidler: Til HGPS er der i de senere år udviklet målrettede behandlinger. Et eksempel er farnesyltransferase‑inhibitoren lonafarnib, som har vist forbedring i overlevelse og hjerte-kar-parametre hos nogle patienter. Der forskes desuden i antisense‑oligonukleotider, småmolekyle‑hæmmere og genterapi.
- Symptomatisk støtte fra fysioterapi, ergoterapi, tandpleje, psykologisk støtte og socialrådgivning.
Genetisk rådgivning
Genetisk rådgivning er vigtig for familier. For nogle syndromer (autosomalt recessive) er der en gen‑bærer‑risiko for søskende; for HGPS er de fleste tilfælde sporadiske (de novo), så tilbagevendelsesrisiko er lav, men rådgivning afhænger af den konkrete genetiske årsag.
Prognose og forskning
Prognosen afhænger af typen af progeroid syndrom. HGPS fører ofte til tidlig død i barne- eller teenagealderen pga. fremskreden åreforkalkning, mens Werner typisk medfører sygdomsproblemer i voksen alder med kortere forventet levetid pga. kræft og kardiovaskulære komplikationer. Mange af disse sygdomme er meget sjældne.
Forskningen i progeroide syndromer er aktiv, fordi de giver unikke indblik i aldringsmekanismer, cellebiologi og DNA‑stabilitet. Studier af progerin, lamin‑funktion og DNA‑reparationsveje har potentiale til at forbedre forståelsen af både sjældne sygdomme og normal aldring, og kan føre til nye behandlingsstrategier for både progeroide patienter og aldersrelaterede sygdomme generelt.
Spørgsmål og svar
Q: Hvad er progeroide syndromer (PS)?
A: Progeroide syndromer er en række genetiske lidelser, hvor den ramte person ser ud til at blive ældre hurtigere.
Q: Hvad forårsager PS?
A: De fleste kendte PS-mutationer fører enten til defekter i DNA-reparationsmekanismen eller defekter i et protein kendt som lamin A/C.
Q: Hvad betyder progeroid?
A: Progeroid betyder at ligne alderdom.
Q: Kan Alzheimers og Parkinsons sygdomme betragtes som progeroide syndromer?
A: Nej, Alzheimers og Parkinsons sygdomme påvirker kun ét væv, og udtrykket progeroidt syndrom bruges om tilfælde, hvor de berørte personer kun viser nogle af aldringens træk, men ikke dem alle.
Q: Hvor mange forskellige typer væv kan være påvirket ved progeroide syndromer?
A: Ved progeroide syndromer er mange forskellige slags væv påvirket.
Q: Hvad er levetiden for personer med PS-relaterede lidelser?
A: Personer med PS-relaterede lidelser har ofte en kortere levetid.
Q: Hvad er de mest undersøgte progeroide syndromer, og hvorfor?
A: De mest undersøgte progeroide syndromer er Werners syndrom (WS) og Hutchinson-Gilford Progeria Syndrome (HGPS), fordi de ligner naturlig aldring.
Relaterede artikler
Forfatter
AlegsaOnline.com Progeroide syndromer (progeria): årsager, symptomer og genetik Leandro Alegsa
URL: https://da.alegsaonline.com/art/79367
Kilder
- doi.org : 10.1093/hmg/ddl214
- pubmed.ncbi.nlm.nih.gov : 16987878