Hæmofili – symptomer, årsager, arvegang og behandling
Hæmofili – forstå symptomer, årsager, arvegang og moderne behandlinger. Lær om typer, diagnostik, blødningsrisiko og behandlingsmuligheder for patienter.
Hæmofili er en blodsygdom, som betyder, at blødningen kan vare meget længere end normalt, fordi blodet ikke størkner som det skal. Personer med hæmofili mangler eller har lave niveauer af bestemte koagulationsfaktorer (blodproteiner), som hjælper med at danne en blodprop. Selv et lille snit eller en indre blødning (se f.eks. blå mærker) kan være farlig — i ekstreme tilfælde kan en stor blødning føre til, at man forbløder til døde. Ordet hæmofili kommer af de græske ord haima ("blod") og philia ("at elske").
Billedgalleri
3 Billeder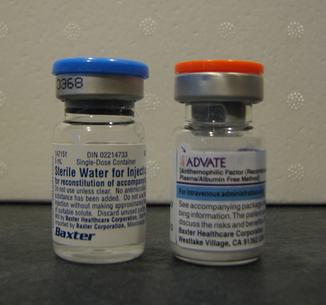

Typer og hvor almindeligt det er
- Hæmofili A (manglende eller nedsat faktor VIII) udgør ca. 90 % af tilfældene. Sværhedsgraden varierer fra mild til svær afhængig af hvor meget faktor VIII der er til stede.
- Hæmofili B (manglende eller nedsat faktor IX) er mindre almindelig end A, men kan være lige så alvorlig. Sværhedsgraden varierer også her.
- Hæmofili C skyldes typisk mangel på faktor XI. Denne form er ofte mildere og arves på anden vis (ofte autosomal recessiv).
Forekomst: Hæmofili A ses i cirka 1 ud af 5.000–10.000 mandlige fødsler; hæmofili B i cirka 1 ud af 20.000–34.000 mandlige fødsler.
Årsag og arvegang
Hæmofili skyldes ændringer i de gener, der koder for koagulationsfaktorer. Særligt ved hæmofili A og B er det ofte fejl på X-kromosomet: Genetiske defekter på X-kromosomet rammer oftere mænd, fordi mænd kun har ét X-kromosom. En kvinde har to X-kromosomer, og et normalt gen på det andet X kan ofte kompensere for et sygt gen, så kvinder oftest er bærere uden svære symptomer. Det er denne type arv man i genetik kalder kønsbundet (X-bunden) arv. Y-kromosomet indeholder få gener og er ikke årsag til disse former for hæmofili. Omtrent 30 % af tilfældene af hæmofili A og B skyldes en ny (de novo) mutation, det vil sige, at der ikke er tidligere tilfælde i familien.
En person med hæmofili omtales ofte som en hæmofilipatient. Kvindelige bærere kan i nogle tilfælde have lavere faktoraktivitet og opleve øget blødningstendens.
Symptomer
Symptomer afhænger af hvor svær sygdommen er (mild, moderat, svær):
- Langvarig blødning efter skrammer, snitsår, tandudtrækning eller operation.
- Spontane blødninger i muskler og led (især knæ, ankler, albuer). Gentagne ledblødninger kan føre til kronisk ledsmerte og ledskade (hæmofili-artropati).
- Større blå mærker (blå mærker) end normalt efter slag eller uden tydelig årsag.
- Næseblod, tandkødsblødning og kraftige menstruationer hos kvinder/bærere.
- Indre blødninger, herunder sjældnere men alvorlige blødninger i hjernen eller indre organer — kræver øjeblikkelig behandling.
Diagnose
Diagnosen stilles via blodprøver, der måler koagulationstider og specifikke faktoraktivitetstal (faktor VIII, IX, XI mv.). Typiske fund kan være forlænget aktiviseret partiel tromboplastintid (aPTT) ved hæmofili A og B. Genetisk testning kan bekræfte mutation og bruges til arvevejledning (fx ved planlægning af graviditet eller undersøgelse af familie).
Behandling
Målet med behandlingen er at stoppe blødninger hurtigt, forebygge blødninger og beskytte led og organer.
- Faktorudskiftning: Den mest effektive behandling er at tilføre manglende koagulationsfaktor (faktor VIII for hæmofili A, faktor IX for B) ved infusion af koncentrater — enten plasmaafledte eller recombinant-produkter. Behandles akut ved blødning og kan gives profylaktisk (fast plan) for at forebygge blødninger, især hos børn med svær hæmofili.
- Desmopressin (DDAVP): Kan øge faktor VIII-niveau midlertidigt hos mange med mild til moderat hæmofili A og bruges ved mindre blødninger eller før mindre indgreb.
- Antifibrinolytika: Medicin som tranexamsyre eller aminokapronsyre hjælper med at stabilisere blodpropper ved f.eks. tandbehandling eller slimhindeblødninger.
- Behandling af inhibitorer: Nogle patienter udvikler antistoffer (inhibitorer) mod tilførte faktorer. Det kræver særlige behandlinger som bypass-agenter (fx rekombinant aktiv faktor VII) eller immun tolerance induktion.
- Nyere tiltag: Genetisk genterapi har vist lovende resultater i kliniske forsøg og kan på sigt give længerevarende eller permanent forbedring af faktorproduktionen for nogle patienter.
Komplikationer
- Kroniske ledsmerter og ledskader efter gentagne blødninger.
- Udvikling af inhibitorer mod behandlingsfaktorer, som gør behandling vanskeligere.
- Alvorlige indre blødninger, herunder blødning i hjerne eller organer.
- Blodoverførsel-relaterede risici ved historisk brug af plasma — moderne produkter og screening har reduceret denne risiko betydeligt.
Praktiske råd og akut håndtering
- Ved mindre blødning: tryk direkte på såret, løft ulykkesområdet og søg lægehjælp hvis blødningen fortsætter. Brug gerne is for at reducere hævelse.
- Ved alvorlig eller vedvarende blødning: kontakt akut lægehjælp eller blødningsklinik; behandling med faktor er ofte nødvendig.
- Undgå NSAID-præparater (fx ibuprofen, aspirin) da de øger blødningstendensen; paracetamol er normalt mere sikkert til smertebehandling.
- Mundhygiejne og forsigtighed ved tandbehandling — ofte nødvendig med præventiv faktorbehandling ved større tandkirurgi.
- Vaccinationer, sportsanbefalinger og kirurgisk planlægning bør ske i samarbejde med specialister i hæmostase.
Graviditet, fødsel og familieplanlægning
Kvinder, der er bærere, bør have rådgivning før og under graviditet. Særlige overvejelser gælder for fødsel og nyfødtpleje, fordi drenges risiko for at være syge er forøget ved X-bunden arvegang.
Søg hjælp og opfølgning
Hæmofili kræver ofte tværfaglig opfølgning på et specialcenter med adgang til hæmatologer, fysioterapeuter og andre relevante faggrupper. Med moderne behandling kan de fleste leve aktive liv, men tidlig diagnose, korrekt behandling og forebyggelse af ledskader er afgørende.
Arv og gener: Sygdommen overføres oftest fra mor til barn gennem generne, hvilket forklarer den hyppigere forekomst hos mænd. Der findes ingen universel helbredelse i dag, men der findes effektive behandlinger og løbende forskning, som forbedrer prognosen for mange berørte.

Relaterede sider
Spørgsmål og svar
Sp: Hvad er hæmofili?
A: Hæmofili er en blodsygdom, der betyder, at blødningen ikke stopper. Blødningen opstår, fordi blodet ikke størkner på grund af mangel på proteiner i blodet, som danner skorper og blodpropper.
Spørgsmål: Hvordan behandles hæmofili?
Svar: Hæmofili kan behandles ved at få en bloddonation fra en person uden hæmofili, da deres blod indeholder koagulerende proteiner, som midlertidigt kan lave en normal skorpe.
Spørgsmål: Hvor almindeligt er hæmofili?
A: Hæmofili A forekommer hos ca. 1 ud af 5.000-10.000 mandlige fødsler og hæmofili B hos ca. 1 ud af 20.000-34.000 mandlige fødsler.
Spørgsmål: Er der nogen kur mod denne sygdom?
A: Der findes ingen kur mod denne sygdom, men der findes forskellige behandlinger rundt om i verden.
Spørgsmål: Hvad er årsagen til hæmofili?
A: Hæmofili rammer normalt mænd, og den overføres fra mor til barn gennem gener. 30 % af hæmofili A- og B-tilfældene er den første person i deres familie, der får hæmofili, hvilket er resultatet af en uventet mutation (det betyder, at der sker en uventet ændring i kroppen). Det rammer normalt mænd på grund af genetiske defekter på X-kromosomet, da de kun har ét X-kromosom, mens kvinder har to X-kromosomer, så et recessivt gen kan være maskeret af et normalt gen på det andet X-kromosom. Defekter af denne type kaldes "kønsrelaterede" i genetik.
Spørgsmål: Er der forskellige typer hæmofilier?
A: Ja, der findes tre typer hæmofelia - hæmofelia A (ingen koagulationsevne), hæmofelia B (ikke tilstrækkelig koagulationsevne) og hæmofelia C (forårsaget af to recessive gener).
Relaterede artikler
Forfatter
AlegsaOnline.com Hæmofili – symptomer, årsager, arvegang og behandling Leandro Alegsa
URL: https://da.alegsaonline.com/art/41733
Kilder
- etymonline.com : "Online Etymology Dictionary"
- hemophilia.org : "Hemophilia B"