Thalassæmi – hvad er det? Årsager, symptomer og behandling
Thalassæmi – forstå årsager, symptomer og behandling: arv, diagnose, komplikationer og muligheder som jernhåndtering og knoglemarvstransplantation. Få klar, praktisk vejledning.
Thalassæmi (eller thalassæmi) er en genetisk sygdom i blodet, som stammer fra Middelhavsområdet.
Denne sygdom skyldes svækkelse og ødelæggelse af de røde blodlegemer. Det skyldes muterede gener, som påvirker kroppens måde at lave hæmoglobin på. Hæmoglobin er det protein i de røde blodlegemer, som transporterer ilt. Personer med thalassæmi danner mindre hæmoglobin og færre cirkulerende røde blodlegemer end normalt, hvilket resulterer i mild eller alvorlig anæmi.
Thalassæmi kan forårsage betydelige komplikationer, herunder lungebetændelse, jernoverbelastning, knogledeformiteter og hjerte-kar-sygdomme. Denne arvelige sygdom i de røde blodlegemer giver imidlertid en vis beskyttelse mod malaria, som er eller var almindelig i de regioner, hvor arten er almindelig. Denne selektive overlevelsesfordel for bærere (kendt som heterozygote fordele) er ansvarlig for at holde mutationen i populationer langt over dens mutationsrate. Bærere er heterozygote for thalassemia-allelen, hvilket betyder, at kun den ene af deres to alleler er mutant (unormal). Der findes en række forskellige versioner af thalassæmi. Hver af dem skyldes en mutation i en anden position i genomet.
I den henseende ligner thalassæmi en anden genetisk sygdom, der påvirker hæmoglobin, nemlig seglcellesygdom.
Det er muligt at helbrede patienter med thalassæmi med knoglemarvstransplantationer fra kompatible donorer. Denne metode kræver dog en HLA-matchet kompatibel donor.
Billedgalleri
8 Billeder



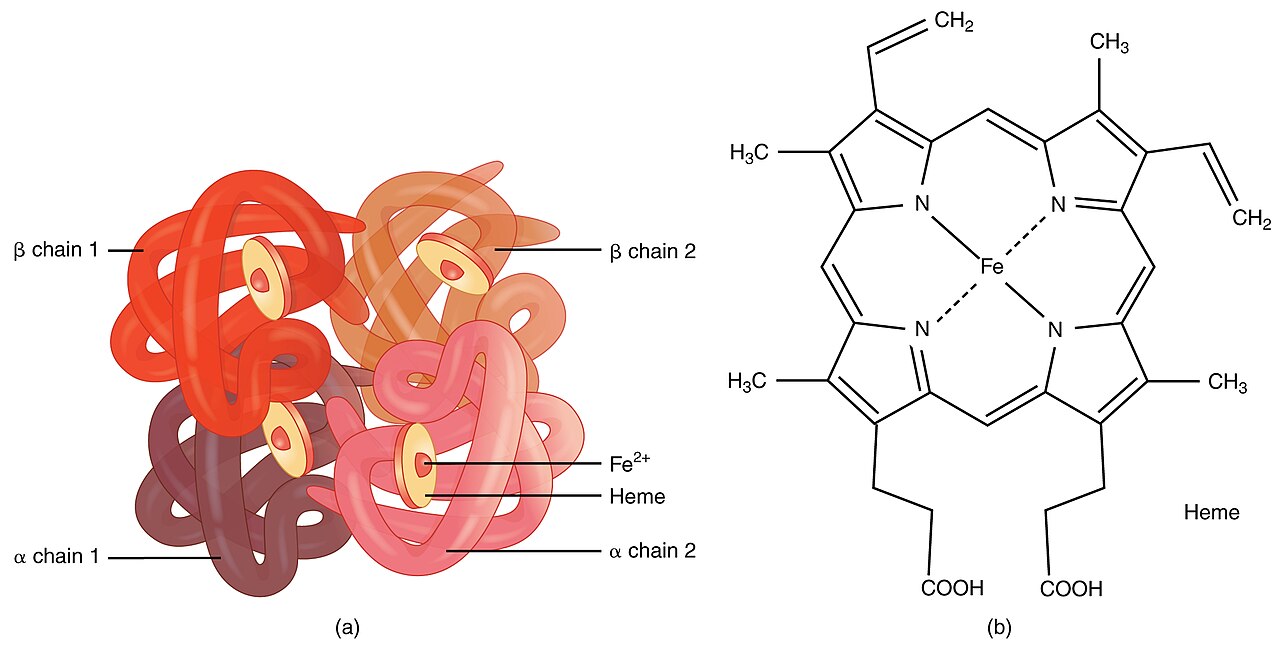
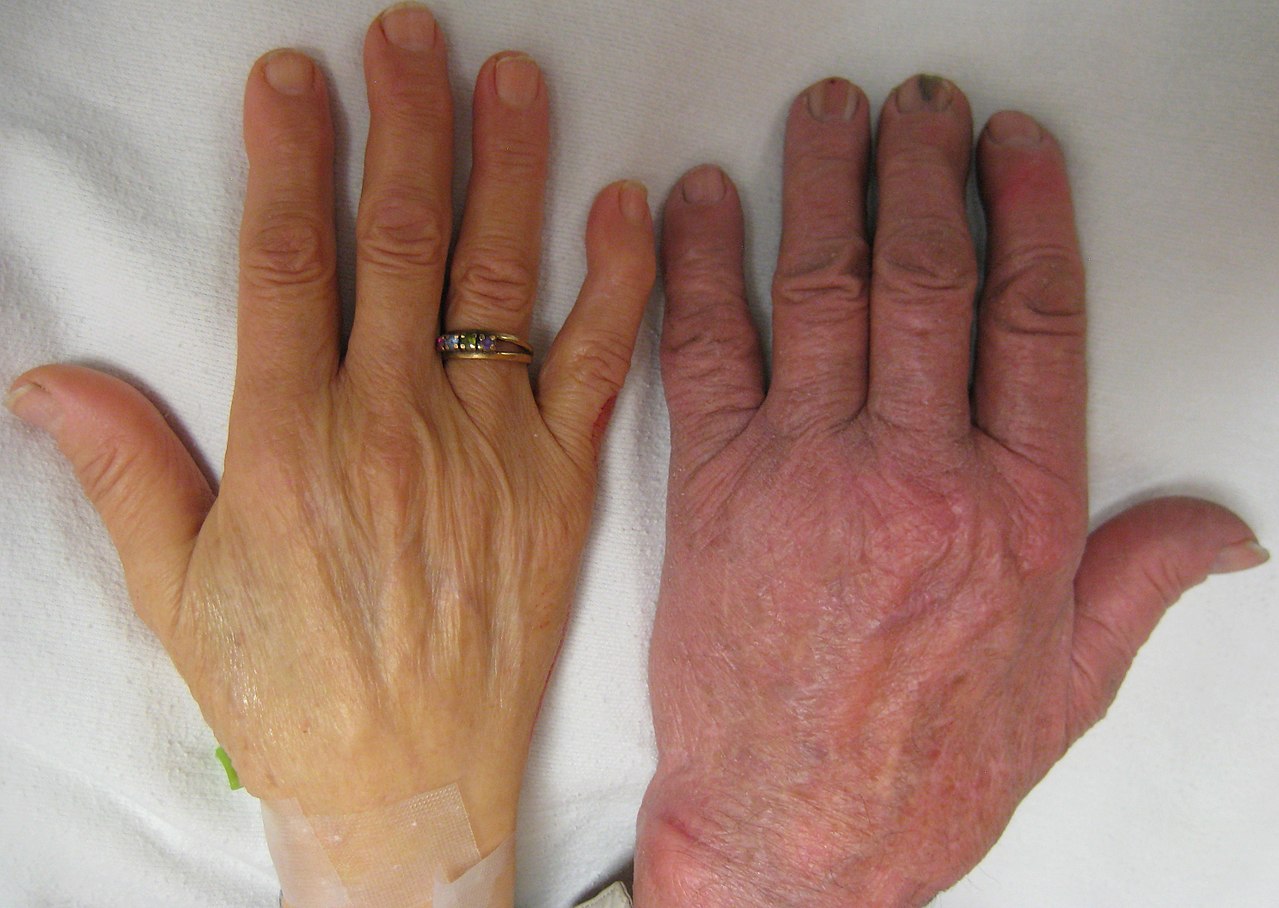


Typer af thalassæmi
Der er to hovedformer:
- Beta-thalassæmi – skyldes mutationer i generne for beta-kæderne i hæmoglobin. Er almindelig i Middelhavet, Mellemøsten, Sydasien og Nordafrika.
- Alfa-thalassæmi – skyldes mutationer i alfa-kæde-generne. Ses især i Sydøstasien, Afrika og nogle dele af Middelhavet.
Inden for disse grupper findes varianter efter sværhedsgrad: bærertilstand (thalassæmi minor/trait), thalassæmi intermedia og thalassæmi major (ofte kaldet Cooley-anæmi), som er den mest alvorlige form.
Arv og genetisk forklaring
Thalassæmi arves typisk autosomalt recessivt: en person med to sygdomsalleler (homozygot) får som regel en alvorlig form, mens en person med én muteret og én normal allel (heterozygot) ofte er bærer med få eller ingen symptomer. Kombinationen af forældrenes gener bestemmer risikoen for, at et barn får sygdommen.
Symptomer og tegn
Symptomer afhænger af sværhedsgraden:
- Carriertilstand (trait): ofte ingen eller meget mild anæmi. Mange opdages kun ved blodprøver.
- Thalassæmi intermedia: moderat anæmi, træthed, blege, nogle gange kræver lejlighedsvis blodtransfusion.
- Thalassæmi major: svær anæmi, træthed, manglende vækst hos børn, gulsot, forstørret milt og lever, knogleforandringer (især i ansigt og kranie) som følge af øget knoglemarvsaktivitet.
Typiske blodprøvefund er microcytær hypokrom anæmi (lavt MCV, lavt MCH), variable røde blodlegemer, og ved beta-thalassæmi ofte ændret fordeling af hæmoglobinfraktioner (fx forhøjet HbA2). Diagnosen bekræftes med hæmoglobinelektroforese og genetisk testning.
Komplikationer
- Jernoverbelastning: især ved gentagne blodtransfusioner eller som følge af øget jernabsorption. Jern ophobes i hjerte, lever og endokrine kirtler og kan give hjertesvigt, leverskade og hormonforstyrrelser.
- Infektioner: patienter, der har fået fjernet milten (splenektomi), har øget risiko for alvorlige infektioner med kapselbærende bakterier.
- Hjerte-kar-sygdomme: skyldes både jernoverbelastning og kronisk anæmi.
- Vækst- og udviklingsproblemer: især hos ubehandlede børn med alvorlig sygdom.
Diagnose
Diagnosen stilles ved kombination af:
- Blodprøver (hæmatologi): Hb, MCV, MCH, antal røde blodlegemer.
- Hæmoglobinelektroforese eller HPLC for at bestemme hæmoglobinvarianter (fx HbA2, HbF).
- Genetisk testning for at påvise specifikke mutationer i alfa- eller beta-globin-generne.
- Prænatal diagnostik (CVS eller fostervandsprøve) kan bruges ved kendt risiko i familien.
Behandling
Behandlingen afhænger af typen og sværhedsgraden:
- Regelmæssige blodtransfusioner – standard for thalassæmi major for at opretholde tilstrækkeligt hæmoglobinniveau og forhindre væksthæmning og knogledeformiteter.
- Jernkatalysator (jernchelation) – nødvendig ved gentagne transfusioner for at fjerne overskydende jern. Almindelige chelatorer er deferoxamin (injektion/infusion), deferasirox og deferipron (orale). Overvågning med serumferritin og lever- og hjertescanning anbefales.
- Splenektomi – kan overvejes ved stor miltforstørrelse eller hyppigt transfusionsbehov, men øger infektionsrisiko og kræver vaccinationer og evt. profylakse.
- Knoglemarvstransplantation (allogen stamcelletransplantation) – den eneste etablerede kurative behandling i dag, men kræver en HLA-matchet donor og medfører risiko for komplikationer som graft-versus-host-sygdom (GVHD).
- Nyere behandlingsmuligheder: genteknologi og genmodificerede stamcelleteknikker (eksempelvis lentiviral genaddition eller CRISPR-baserede metoder) viser lovende resultater i kliniske forsøg og kan i fremtiden tilbyde kurative alternativer for flere patienter.
- Støttende behandling: vaccination mod pneumokokker, meningokokker og Haemophilus influenzae b ved splenektomi; behandling af infektioner; væksthormon ved behov; og specialiseret graviditetsomsorg ved påvirkede kvinder.
Forebyggelse og genetisk rådgivning
På grund af arvegangen er genetisk rådgivning vigtig for berørte familier. Bærertestning, prækonceptionel rådgivning, prænatal diagnostik og muligvis præimplantationsdiagnostik (PGD) ved IVF kan hjælpe par med at træffe informerede valg. I risikoområder tilbydes ofte screeningsprogrammer.
Prognose
Prognosen er forbedret dramatisk med moderne transfusionsregimer, jernchelatering og specialiseret pleje. Personer med thalassæmi minor lever normalt et fuldt liv uden behandling. For patienter med thalassæmi major kan god behandling give mange års overlevelse og bedre livskvalitet; helbredelse er mulig med vellykket stamcelletransplantation eller kommende genterapier.
Praktiske råd
- Søg specialistvurdering hos hæmatolog ved mistanke om thalassæmi.
- Hvis du er bærer eller har familiær risiko, få genetisk rådgivning før graviditet.
- Ved regelmæssige transfusioner: følg anbefalinger om jernmåling og chelation for at beskytte hjerte og lever.
- Hav styr på vaccinationer, især hvis fjernet milt.
Spørgsmål og svar
Q: Hvad er thalassæmi?
A: Thalassæmi er en genetisk blodsygdom, som stammer fra Middelhavsområdet. Sygdommen skyldes, at de røde blodlegemer svækkes og ødelægges på grund af muterede gener, der påvirker den måde, hvorpå kroppen laver hæmoglobin.
Spørgsmål: Hvilke komplikationer er der forbundet med thalassæmi?
A: Komplikationer i forbindelse med thalassæmi kan omfatte lungebetændelse, jernoverbelastning, knogledeformiteter og hjerte-kar-sygdomme.
Spørgsmål: Hvordan beskytter thalassæmi mod malaria?
A: Bærere af thalassæmi har en selektiv overlevelsesfordel for bærere (kendt som heterozygote fordele), hvilket bidrager til at holde mutationen i populationer langt over dens mutationsrate. Dette giver dem beskyttelse mod malaria, som er eller var almindelig i regioner, hvor dette træk er almindeligt forekommende.
Spørgsmål: Er der forskellige versioner af thalassæmi?
Svar: Ja, der findes en række forskellige versioner af thalassæmi, som hver især skyldes en mutation i en anden position i genomet. Den ligner en anden genetisk sygdom, der påvirker hæmoglobin, nemlig seglcellesyge.
Spørgsmål: Er det muligt at helbrede patienter med thalassæmi?
Svar: Ja, det er muligt at helbrede patienter med thalassæmi ved hjælp af knoglemarvstransplantationer fra kompatible donorer, som har en HLA-matchet kompatibel donor.
Relaterede artikler
Forfatter
AlegsaOnline.com Thalassæmi – hvad er det? Årsager, symptomer og behandling Leandro Alegsa
URL: https://da.alegsaonline.com/art/97356
Kilder
- accessmedicine.com : accessmedicine.com/content.aspx?aID=6123722
- mayoclinic.com : mayoclinic.com/health/thalassemia/DS00905/DSECTION=complications
- bloodjournal.hematologylibrary.org : HLA-matched sibling bone marrow transplantation for β-thalassemia major