Cystisk fibrose (mukovisidose): arvelig sygdom — symptomer og behandling
Cystisk fibrose (mukovisidose): arvelig sygdom med tykt slim — symptomer, arvegang og moderne behandlingsmuligheder.
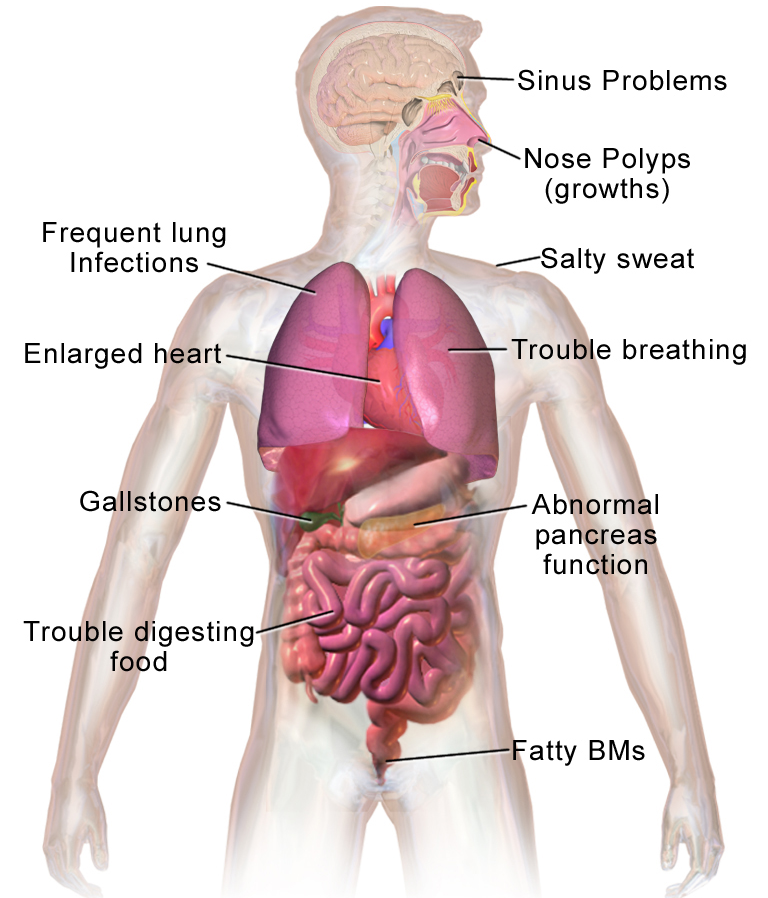
Cystisk fibrose, også kaldet mucoviscidose eller forkortet CF, er en arvelig sygdom, hvor kroppens kirtler producerer tykt, klæbrigt slim. Slimet kan ophobes i lungerne, i fordøjelsessystemet og i andre organer, hvilket giver kroniske luftvejsinfektioner, problemer med fordeling og optagelse af madstoffer og andre følgetilstande. sygdommen smitter ikke fra person til person.
Billedgalleri
4 Billeder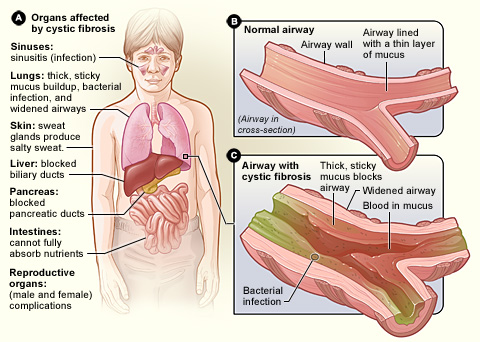

Arvelighed og gener
Cystisk fibrose skyldes skader i det såkaldte CFTR-gen. For at et barn skal få sygdommen, skal det arve et sygdomstilfredsstillende gen fra begge forældre (autosomalt recessiv arv). Det betyder, at forældre kan være raske bærere af et forandret CFTR-gen uden selv at have symptomer. Hvis begge forældre er bærere, er risikoen for, at et barn får CF, 25 % pr. graviditet. Der findes mange forskellige mutationer i CFTR-genet, og typen af mutation påvirker både sygdomsmanifestation og hvilke behandlinger, der virker.
Symptomer og typiske problemer
Symptomer kan variere meget i sværhedsgrad og debutalder, men almindelige problemer er:
- Lunger: Gentagne luftvejsinfektioner, kronisk hoste, piben, nedsat lungefunktion og arvævsdannelse (bronchiectasier). Hyppige bakterier omfatter Staphylococcus aureus, Pseudomonas aeruginosa og i nogle tilfælde Burkholderia-arter.
- Fordøjelse: Pancreas (bugspytkirtlen) kan være påvirket, hvilket giver dårlig fordøjelse af fedt og proteiner, tynd afføring, oppustethed og manglende vægtøgning hos børn (pancreasinsufficiens).
- Næringsstoffer: Malabsorption kan føre til vækstproblemer og mangel på fedtopløselige vitaminer (A, D, E og K).
- Andet: Saltmangel og dehydrering i varme perioder, næsepolypper, kronisk bihulebetændelse, diabetes relateret til CF (CFRD) og hos de fleste mænd fravær af den sædledende kanal (infertilitet).
Diagnose
CF kan opdages via nyfødtscreening (måling af immunoreaktivt trypsinogen - IRT) i mange lande, blodprøver og gentests. Den klassiske bekræftende test er svedtest, hvor man måler saltindholdet i sveden (øget ved CF). Genetisk test kan identificere specifikke CFTR-mutationer og afgøre egnetheden for målrettede behandlinger.
Behandling
Der findes i dag ingen fuldstændig kur, men moderne behandlinger gør det muligt at leve længere og have bedre livskvalitet. Behandling er tværfaglig og foregår ofte på specialcentre, hvor læger, fysioterapeuter, diætister og andet sundhedspersonale samarbejder.
Vigtige elementer i behandlingen:
- Airway clearance (lungerydding): Fysioterapi og teknikker til at løsne og fjerne slim (fx PEP-fløjte, fysisk træning, doulas og andre manuelle teknikker).
- Inhalationsbehandlinger: Bronkodilatatorer, slimløsende medicin som dornase alfa (Pulmozyme) og hypertonisk saltvand, som hjælper med at gøre sekretet mindre tykt.
- Antibiotika: Til behandling og forebyggelse af infektioner; kan gives som tabletter, inhalation eller intravenøst ved svære infektioner. Langvarige eller gentagne infektioner kan kræve specialtilpasset antibiotikastrategi.
- CFTR-modulatorer: Nyere lægemidler (fx ivacaftor, kombinationer som lumacaftor/ivacaftor, tezacaftor/ivacaftor og trippel-kombinationer med elexacaftor/tezacaftor/ivacaftor) retter sig mod selve det defekte CFTR-protein og kan betydeligt forbedre lungefunktion og ernæring hos patienter med visse mutationer. Disse behandlinger virker kun for bestemte genfejl, så genetisk analyse er nødvendig for at afgøre, hvem der har gavn af dem.
- Pancreasenzymer og kost: Pancreasenzymer til måltider, energirig kost og tilskud af fedtopløselige vitaminer er vigtige for at sikre vækst og ernæring.
- Behandling af komplikationer: Behandling af CF-relateret diabetes, næsepolypper og kroniske bihuleproblemer efter behov. I fremskredne tilfælde kan lungetransplantation være en mulighed.
Sundhedspleje og forebyggelse
Personer med CF har gavn af regelmæssig opfølgning på et center med erfaring i CF. Forebyggende tiltag omfatter:
- Vaccinationer (influenza, pneumokok m.fl.).
- God håndhygiejne og infektionskontrol for at reducere risiko for smitte med skadelige bakterier.
- Øget opmærksomhed på væske- og saltbalance, særligt hos små børn og i varmt klima.
- Fysisk aktivitet og træning for at styrke lunger og almentilstand.
Prognose
Prognosen for mennesker med cystisk fibrose er forbedret markant over de sidste årtier på grund af bedre behandling og nye lægemidler. Forventet levetid varierer, men mange patienter lever godt ind i voksenalderen og op i 30'erne, 40'erne og længere afhængigt af sygdommens sværhedsgrad, adgang til specialiseret behandling og brug af moderne lægemidler.
Genetisk rådgivning
Hvis der i familien er cystisk fibrose eller man har mistanke om bærerstatus, kan genetisk rådgivning give information om risiko ved graviditet og muligheden for sygdoms- eller bærertest. Prenatal diagnostik og præimplantationsgenetisk diagnostik (PGD) er muligheder for par, der ønsker det.
Opsummering: Cystisk fibrose er en arvelig, ikke-smitsom sygdom, der påvirker slimproduktionen og især rammer lunger og fordøjelse. Der findes ingen kur, men moderne behandlinger — herunder nye målrettede lægemidler — kan forbedre symptomer, hæmme sygdomsudvikling og øge livskvalitet og forventet levetid. Tidlig diagnose, tværfaglig opfølgning og genetisk rådgivning er centrale elementer i plejen.
Hvad CF gør ved kroppen
Cystisk fibrose påvirker hele kroppen. Overordnet set har kroppen problemer med at flytte salt til de dele af kroppen, der har brug for det. Da kroppen har problemer med at flytte salt, hober det sig op på steder, hvor det ikke skal være, f.eks. i lungerne, maven og tarmene.
Lunger
Når salt sætter sig fast i lungerne, medfører det, at der bliver mindre vand i lungerne, hvilket gør, at slim bliver meget tykt. Det bliver meget svært at trække vejret. Behandlingen af dette er indåndingsmedicin, der hjælper med at tilføre vand til lungerne for at holde slimet tyndere, så det er lettere at hoste op. Når der er tyndere og mindre slim, er det lettere at trække vejret.
Behandling
Der findes ingen kur mod cystisk fibrose. Selv om folk kan gøre noget for at holde sig raske. Sunde vaner holder personen fra at blive mere syg. Folk kan holde sig rene. Folk kan holde sig væk fra bakterier. De kan drikke vand for at hjælpe slimen til at forsvinde. At tage enzymer hjælper med at fordøje maden, hvis der er slim i maven.
Motion fjerner slim. Det opbygger stærke muskler og knogler og styrker lungerne. Indtagelse af vitaminer hjælper kroppen med at bekæmpe infektionen. Det hjælper også kroppen med at vokse og fungere godt.
- Indåndingsantibiotika bruges til at forhindre bakterier i at vokse i det tykke slim.
- Inhaleret saltvand hjælper med at holde lungerne fugtige
Test for cystisk fibrose
- Svedkloridtest - denne test tester saltniveauet i en persons sved.
- Genetisk test - denne test anvendes, hvis svedprøven er positiv, for at se, om de har begge gener.
65 roser
"65 roser" er den måde, som nogle børn omtaler deres tilstand på, da cystisk fibrose er svært for et lille barn at sige. "65 roser" er også et varemærkebeskyttet udtryk, som Cystic Fibrosis Foundation har taget i brug for at kontrollere brugen af det. Det er en meget nyttig måde for små børn at forstå det på. Når det tales højt, lyder det på samme måde som cystisk fibrose.
Spørgsmål og svar
Q: Hvad er cystisk fibrose?
A: Cystisk fibrose er en tilstand, der får kroppen til at producere tykt, klæbrigt slim, der kan ophobe sig i lungerne, fordøjelsessystemet og andre dele af kroppen.
Q: Hvordan opstår cystisk fibrose?
A: Cystisk fibrose skyldes, at man arver genet for cystisk fibrose fra begge forældre.
Q: Kan en person med kun ét cystisk fibrose-gen give sygdommen videre til sit barn?
A: En person med kun ét cystisk fibrose-gen har måske ikke selv sygdommen, men kan stadig give genet videre til sit barn.
Q: Er cystisk fibrose smitsom?
A: Nej, cystisk fibrose er ikke smitsom og kan ikke overføres fra en person til en anden.
Q: Findes der en kur mod cystisk fibrose?
A: Nej, der findes i øjeblikket ingen kur mod cystisk fibrose, men der er mange medikamenter, der kan hjælpe med at håndtere tilstanden.
Q: Hvilke dele af kroppen kan blive påvirket af cystisk fibrose?
A: Cystisk fibrose kan påvirke lungerne, fordøjelsessystemet og andre dele af kroppen.
Q: Hvordan kan folk med cystisk fibrose holde sig sunde?
A: Personer med cystisk fibrose kan holde sig sunde ved at tage medicin, overvåge deres tilstand og følge en sund livsstil.
Relaterede artikler
Forfatter
AlegsaOnline.com Cystisk fibrose (mukovisidose): arvelig sygdom — symptomer og behandling Leandro Alegsa
URL: https://da.alegsaonline.com/art/24952