Beregningskemi (computerkemi): metoder, anvendelser og principper
Beregningskemi (computerkemi): metoder, principper og anvendelser — forudsig molekylstrukturer, energier og reaktivitet til design af nye lægemidler og avancerede materialer.
Computerkemi er en gren af kemien, der bruger computervidenskab til at løse kemiske problemer. Disse programmer beregner molekylers og faste stoffers strukturer og egenskaber. Beregningskemi supplerer normalt de oplysninger, der opnås ved kemiske eksperimenter. Den kan forudsige kemiske fænomener, som endnu ikke er blevet observeret. Den anvendes i vid udstrækning til design af nye lægemidler og materialer.
Computerkemi kan forudsige struktur (dvs. de forventede positioner for molekylets atomer), absolutte og relative (vekselvirknings)energier, elektroniske ladningsfordelinger, dipoler og højere multipolmomenter, vibrationsfrekvenser, reaktivitet eller andre spektroskopiske størrelser og tværsnit for kollision med andre partikler.
I computerkemi undersøges både statiske og dynamiske systemer. I alle tilfælde vokser størrelsen af det system, der undersøges, i takt med at computertiden og de andre ressourcer (f.eks. hukommelse og diskplads), der bruges, også. Systemet kan være et enkelt molekyle, en gruppe af molekyler eller et fast stof. Metoderne til beregningskemi spænder fra meget nøjagtige til meget tilnærmede metoder. Meget nøjagtige metoder kan typisk kun anvendes til små systemer.
Billedgalleri
8 Billeder


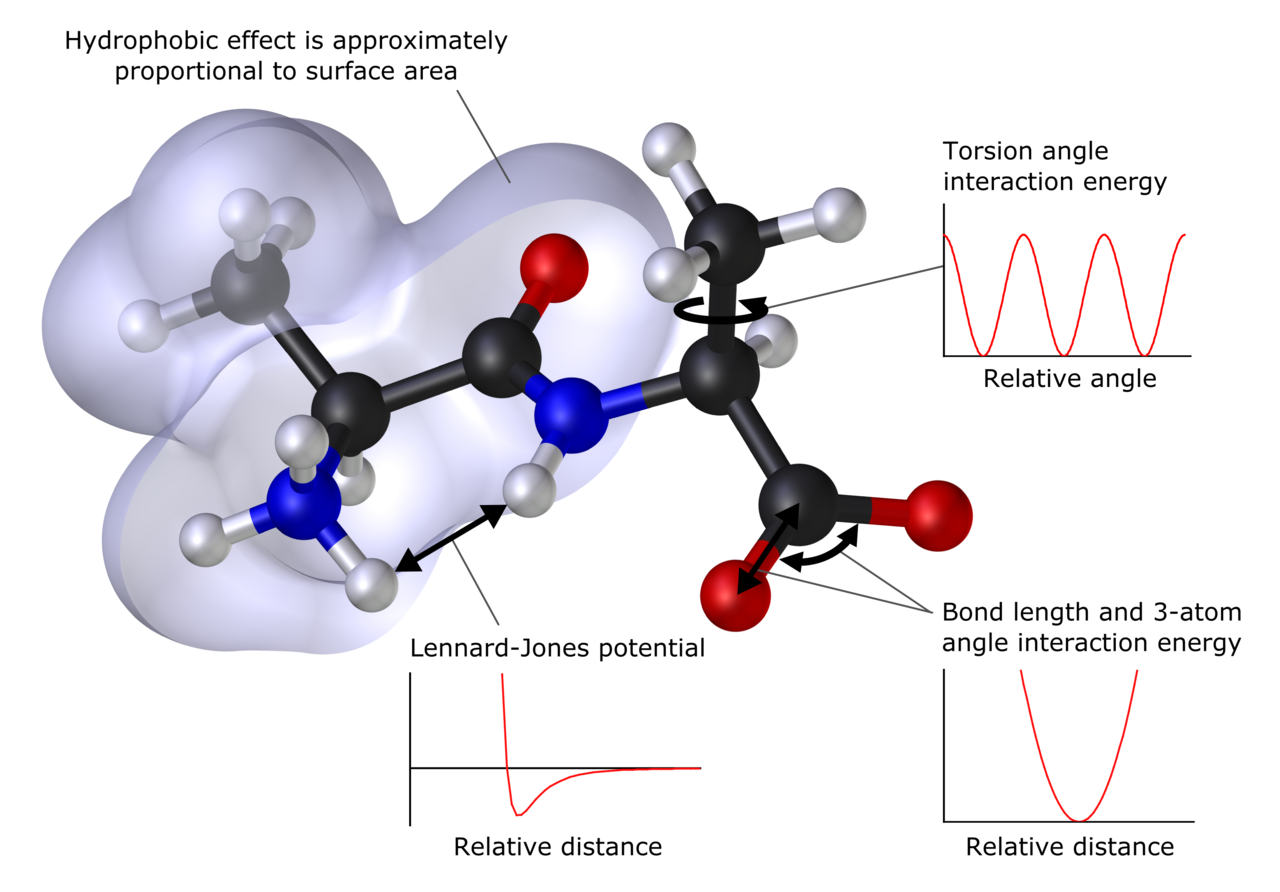


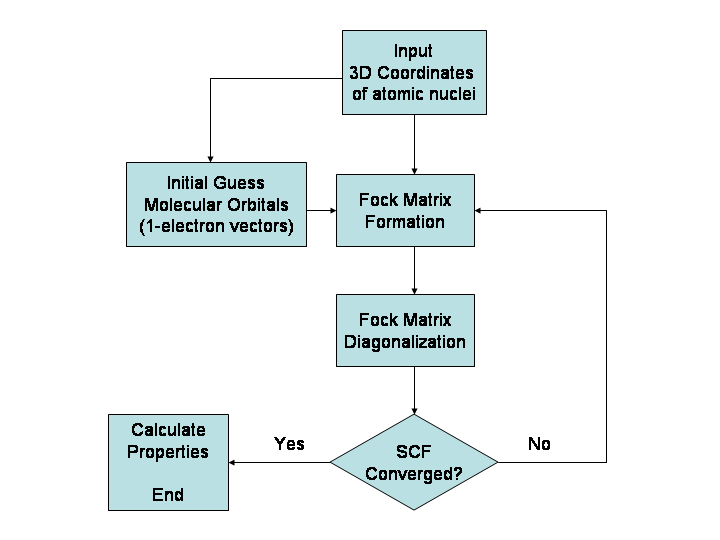
Hovedprincipper og metoder
- Kvantekemiske metoder (ab initio): Beregner elektronernes adfærd ud fra Schrödinger-ligningen uden empiriske parametre. Eksempler er Hartree–Fock (HF) og metoder, der inkluderer korrelation som MP2, CCSD(T). Disse metoder kan være meget præcise, men skalerer hurtigt med systemstørrelsen.
- Density Functional Theory (DFT): Anvender elektron-tætheden som variabel og tilbyder en god balance mellem nøjagtighed og beregningstid for mange systemer. Valget af funktional er afgørende for resultaternes kvalitet.
- Semi-empiriske metoder: Forenkler kvantemekaniske ligninger ved at indføre empiriske parametre. De er hurtigere end ab initio og nyttige til større molekyler, men mindre pålidelige i nye eller uventede kemiske miljøer.
- Molekylmekanik og kraftfelter: Beskriver molekyler ved klassiske potensfunktioner (bondstretch, vinkelbøjning, torsion, ikke-bindende interaktioner). Velegnet til store biomolekyler og materialer, men kan ikke beskrive elektroniske omarrangementer (f.eks. kemiske reaktioner) direkte.
- Hybridmetoder (QM/MM): Kombinerer kvantemekaniske beregninger for den reaktive region med molekylmekanik for resten af systemet. Ofte brugt i enzymkatalyse og i solvatiserede systemer.
- Molekylær dynamik (MD): Simulerer systemers tidsudvikling under klassiske eller kvantemekaniske potentialer for at studere temperaturafhængige og kinetiske fænomener.
Anvendelser
- Lægemiddeldesign: Virtuel screening, bindningsenergiestimering, ledoptimering og forudsigelse af ADMET-egenskaber.
- Materialeforskning: Forudsigelse af krystalstrukturer, elektroniske egenskaber, mekaniske styrker og temperaturstabilitet.
- Katalyse og reaktionsmekanismer: Identifikation af overgangstilstande, aktiveringsenergier og katalytiske cyklusser.
- Spektroskopi: Beregning af NMR-, IR-, UV/Vis- og røntgenspektre for forbindelseidentifikation og tolkning af eksperimenter.
- Miljø- og sikkerhedsanalyse: Forudsigelse af nedbrydningsveje, toksicitet og mobilitet i miljøet.
- Fundamental kemi: Studier af elektronkorrelation, svage interaktioner (van der Waals), og ny kemi, som endnu ikke er syntetiseret eksperimentelt.
Praktiske overvejelser og begrænsninger
- Skaleringsproblemer: Mere præcise metoder kræver ofte kraftigt stigende regnekraft og hukommelse. Valget af metode er derfor en afvejning mellem nøjagtighed og tilgængelige ressourcer.
- Valg af basis sæt og funktional: I kvantemekaniske beregninger påvirker basis sæt og DFT-funktionaler resultaterne markant. Dårligt valg kan give systematiske fejl.
- Solvatisering og temperatur: Omgivelser som opløsningsmiddel eller faste matrikler påvirker resultaterne og kræver særlig behandling (implicit/eksplicit solventmodeller, termostattilknytning i MD).
- Validering: Beregnede resultater bør sammenlignes med eksperimentelle data, når det er muligt, eller med højere-nøjagtighed beregninger for mindre systemer.
- Usikkerheder: Alle beregninger har usikkerheder — både fra teoretiske approksimationer og numeriske implementeringer. Rapportering af estimeret fejlmargin er god praksis.
Software, værktøjer og ressourcer
- Der findes mange kommercielle og åbne kilder til beregningskemi: kvantekemi-pakker, DFT-software, MD-motorer og visualiseringsværktøjer. Valget afhænger af problemets type, størrelse og ønsket nøjagtighed.
- Højtydende beregning på klynger eller cloud-tjenester er ofte nødvendig til større eller mere krævende opgaver.
- Databaser og delte beregningsresultater hjælper med validering og genbrug af metoder og parametre.
God praksis
- Start med enkle metoder og test for konvergens i basis sæt, gradienttolerance og andre numeriske parametre.
- Udfør kontrolberegninger (f.eks. forskellige funktionaler eller basis sæt) for at vurdere følsomhed.
- Dokumentér alle forudsætninger, parametre og versioner af software, så resultater kan reproduceres.
- Samarbejd med eksperimentelle kolleger for at krydstjekke forudsigelser og forbedre modeller.
Computerkemi er et fleksibelt og kraftfuldt felt, der binder teori, numeriske metoder og eksperiment sammen for at give dyb indsigt i kemiske systemer. Ved at vælge rette metode og være opmærksom på begrænsninger kan beregningskemi markant accelerere forskning og udvikling i kemi, biokemi, materialeforskning og beslægtede områder.

Relaterede sider
- Bioinformatik
- Statistisk mekanik
Spørgsmål og svar
Spørgsmål: Hvad er beregningskemi?
Svar: Computerkemi er en gren af kemien, hvor man bruger computervidenskab til at løse kemiske problemer. Den kan bruges til at beregne molekylers og faste stoffers strukturer og egenskaber, forudsige kemiske fænomener, som endnu ikke er blevet observeret, og designe nye lægemidler og materialer.
Spørgsmål: Hvilke typer systemer undersøger computerkemi?
Svar: Beregningskemi undersøger både statiske og dynamiske systemer. Systemet kan være et enkelt molekyle, en gruppe af molekyler eller et fast stof.
Spørgsmål: Hvilke typer oplysninger kan beregningskemi give?
A: Computerkemi kan give oplysninger om f.eks. struktur (atomernes positioner), absolutte og relative energier, elektroniske ladningsfordelinger, dipoler og højere multipolmomenter, vibrationsfrekvenser, reaktivitet eller andre spektroskopiske størrelser og tværsnit for kollisioner med andre partikler.
Spørgsmål: Hvor nøjagtige er de metoder, der anvendes i beregningskemi?
A: Nøjagtigheden af de metoder, der anvendes i beregningskemi, varierer fra meget nøjagtige til meget omtrentlige. Meget nøjagtige metoder kan typisk kun anvendes til små systemer.
Spørgsmål: Hvordan supplerer beregningskemi eksperimentelle data?
A: Beregningskemi supplerer normalt de oplysninger, der opnås ved kemiske eksperimenter. Den kan bruges til at forudsige resultater, som endnu ikke er blevet observeret eksperimentelt.
Spørgsmål: Har størrelsen af det system, der undersøges, indflydelse på, hvor meget computertid der er nødvendig?
A: Ja - efterhånden som størrelsen af det system, der undersøges, vokser, vokser også den mængde computertid, der kræves til analyse, og de ressourcer såsom hukommelse og diskplads, der er nødvendige til lagring.
Relaterede artikler
Forfatter
AlegsaOnline.com Beregningskemi (computerkemi): metoder, anvendelser og principper Leandro Alegsa
URL: https://da.alegsaonline.com/art/22297