Skør knoglesygdom (Osteogenesis imperfecta) – årsager, typer og symptomer
Skør knoglesygdom (OI): årsager, typer og symptomer — forstå genetiske årsager, brudrisiko, syn- og høretab samt muligheder for forebyggelse og behandling.
Osteogenesis imperfecta er en genetisk lidelse, almindeligvis kaldet skør knoglesygdom. Den skyldes oftest ændringer i generne, der koder for kollagen type I — især COL1A1 og COL1A2 — og rammer knoglernes styrke og struktur. Sygdommen kan overføres autosomalt dominant, hvilket betyder, at én syg forælder kan give sygdommen videre til et barn via det unormale gen, men der ses også nye (de novo) mutationer og sjældnere recessive former. Osteogenesis imperfecta blev først beskrevet af Vrolik i 1854, og forekomsten er omtrentlig 1:10.000–1:20.000 fødsler.
Billedgalleri
10 Billeder







Årsager og genetik
De hyppigste årsager er mutationer i generne for kollagen type I (COL1A1 og COL1A2). Kollagen er en vigtig byggesten i knoglevæv — når det er svækket, bliver knoglerne skøre og mere tilbøjelige til brud. I mange tilfælde nedarves sygdommen autosomalt dominant, men der findes også varianter med autosomal recessiv arvegang og nyere beskrevne varianter relateret til andre gener. Nogle personer får sygdommen som følge af en ny mutation uden familiehistorie.
Typer af Osteogenesis imperfecta
Sygdommen er traditionelt inddelt i fire hovedtyper (Sillence-klassifikationen):
- Type I: Den mildeste og hyppigste form. Mange har få brud, normal eller kun let nedsat højde, blålig sklera (øjenskind), og ofte problemer med tænder (dentinogenesis imperfecta). Normal forventet levetid er almindeligvis uændret.
- Type II: Den mest alvorlige form, ofte dødelig i nyfødtperioden eller tidligt spædbarnsår pga. svære knoglebrud og åndedrætsproblemer. Kendetegnes ved mange frakturer prenatalt eller ved fødslen.
- Type III: Svær, deformerende form med mange brud gennem barndommen, væksthæmning, betydelig knogledeformitet og ofte høre- og tandproblemer. Overlevelse til voksen alder er mulig, men med markant funktionsnedsættelse.
- Type IV: Moderat svær form med variabel grad af knogleskørhed, ofte normal eller lidt nedsat sklera og varierende grad af skeletdeformitet.
Der er siden udviklet udvidede klassifikationer med mange genetiske undertyper (over 15–20 genassocierede former), men Sillence-typen bruges stadig som praktisk inddeling ved klinisk vurdering.
Symptomer og kliniske tegn
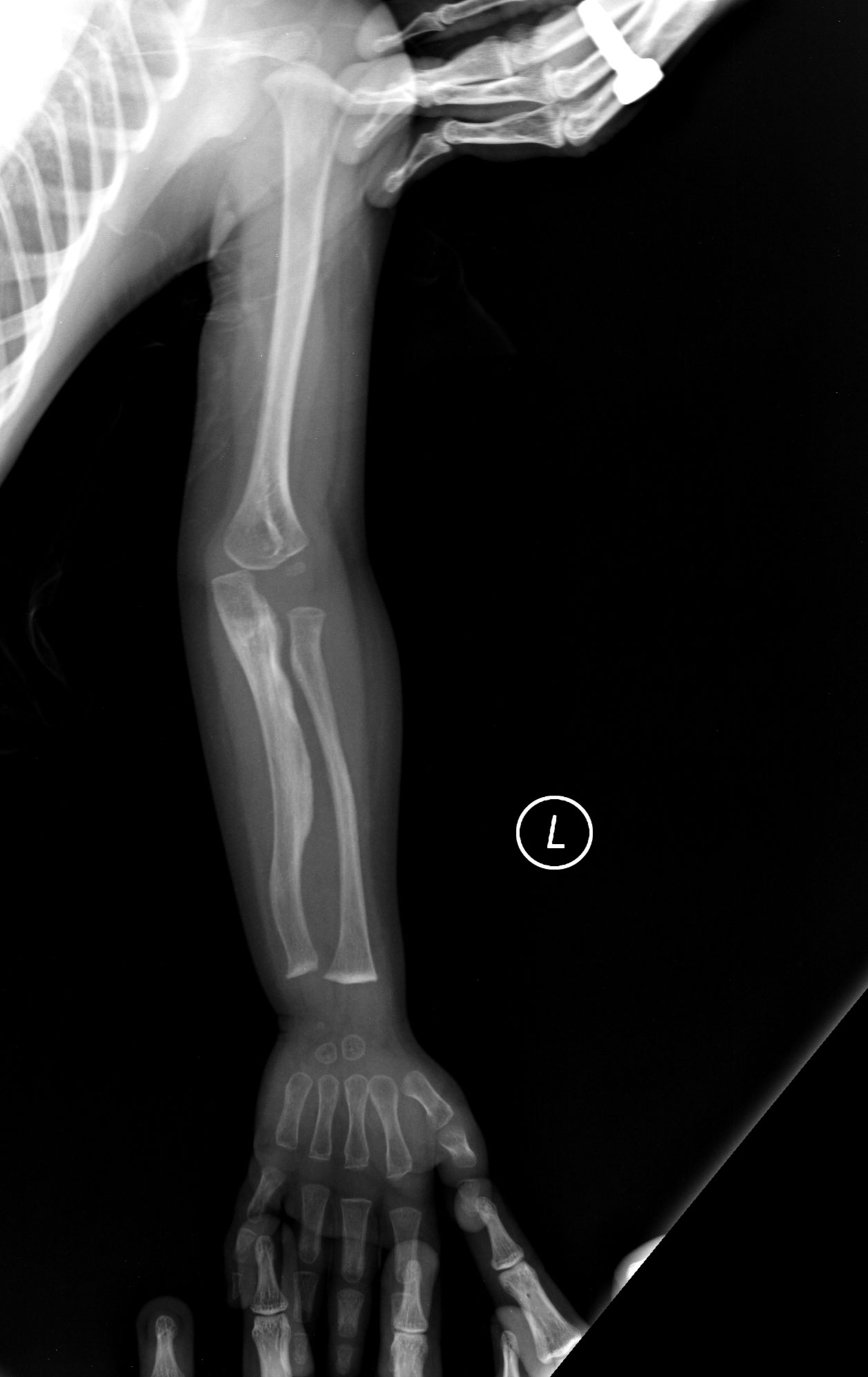
- Mange og gentagne knoglebrud, ofte ved let traume eller spontant.
- Knogledeformiteter (skæv ryg/scoliose, forkortede lemmer, bøjede knogler).
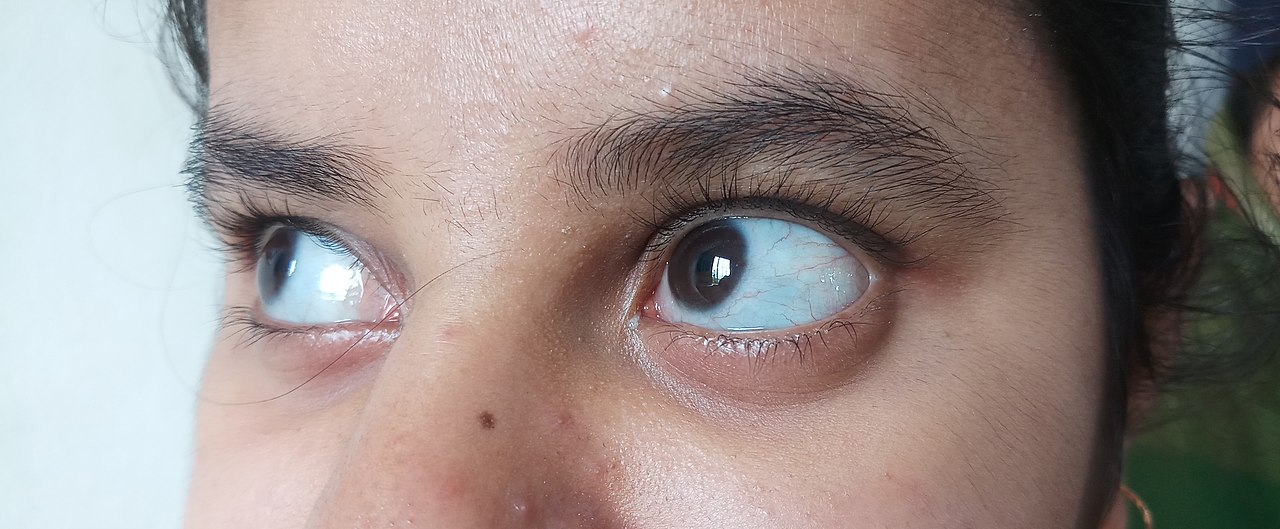
- Blå eller lilla sklera (det hvide i øjet ser blåligt ud) pga. tyndt bindevæv.
- Dentinogenesis imperfecta: skøre, misfarvede eller gennemsigtige tænder, øget slid og kariesfølsomhed.
- Høretab: kan udvikle sig i barndom eller voksenalder og være både konduktivt og sensorineuralt.
- Mindre muskelstyrke, ledhypermobilitet og træthed.
- Påvirket vækst og kortere kropslængde ved sværere former.
- Respiratoriske problemer ved svære thoraxdeformiteter, og i sjældne tilfælde hjerteproblemer.
Diagnose
Diagnosen stilles ud fra kliniske fund (hyppige frakturer, blå sklera, dentale forandringer), røntgenundersøgelser (karakteristiske knogleforandringer), og supplerende undersøgelser som DXA-scanning for knoglemineraltethed. Genetisk testning kan bekræfte årsagen ved at identificere mutationer i relevante gener og kan også bruges til prænatalt eller præimplanteringsdiagnostik (PGD) i familier, der ønsker det.
Behandling og opfølgning
Der findes i dag ingen kurativ behandling, men der er flere effektive tiltag til at reducere symptomer, forebygge komplikationer og forbedre funktion:
- Multidisciplinært team: børnelæger, ortopædkirurger, fysioterapeuter, ergoterapeuter, audiologer og tandlæger (gerne specialiserede i OI).
- Medicinsk behandling med bisfosfonater (fx pamidronat eller zoledronsyre) kan øge knoglemineraltetheden og reducere antal frakturer hos børn og voksne i udvalgte tilfælde.
- Ortopædkirurgi: intramedullære skinner (rodding), korrigerende operationer og stabilisering ved gentagne frakturer eller svære deformiteter.
- Smertestyring, genoptræning og tilpasset fysioterapi for at styrke muskler og øge mobilitet uden at give unødige risici for brud.
- Tandpleje for at håndtere dentinogenesis imperfecta, herunder fyldninger, kroner og forebyggende tandpleje.
- Høreapparater eller operation ved betydeligt høretab; løbende audiologisk opfølgning.
- Kostråd, vitamin D- og calciumtilskud efter behov, og vejledning i aktiviteter for at minimere fald og brud (undgå højrisikosport).
Prognose
Prognosen varierer meget efter type og sværhedsgrad. Type II er ofte perinatalt dødelig, mens type I typisk har normal eller næsten normal levetid med relativt få brud. Type III og IV ligger i mellem og kan give varig funktionsnedsættelse, men mange lever et langt liv med tilpasset behandling og støtte.
Hverdag, graviditet og genetisk rådgivning
Personer med OI kan have et aktivt liv med rette tilpasninger. Ergonomiske hjælpemidler, tilpasset skole- og arbejdsplads, transporthjælpemidler og social støtte er ofte centrale. Ved ønske om børn er genetisk rådgivning vigtig for at gennemgå arvegang, risiko for overførsel og muligheder som prænatalt test eller præimplantationsdiagnostik. Gravide kvinder med OI bør følges tæt af specialistteam, da både graviditet og fødsel kan kræve særlig planlægning.
Når skal man søge læge
Søg lægehjælp ved gentagne brud, pludselig nedsat hørelse, betydelig tandproblematik, vejrtrækningsproblemer eller ved spørgsmål om arvelighed. Et specialiseret center eller team med erfaring i OI kan give bedst mulig vurdering og behandlingstilbud.
Hvis du ønsker videre information, anbefales kontakt til lokale patientforeninger og specialiserede knogle- eller genetiske centre, som kan tilbyde både praktisk støtte og opdateret rådgivning.
Symptomer
Mindre alvorlige symptomer på OI kan omfatte:
- let brækkede knogler
- løse samlinger
- lav muskeltonus
- blå, lilla eller grå farve på den normalt hvide del af øjnene
- trekantet form af ansigtet
- tendens til at udvikle skoliose
- skøre tænder
OI har mange andre alvorlige og dødelige symptomer, herunder åndedrætsbesvær og knogledeformitet.
Demografiske data
OI forekommer i lige høj grad hos både mænd og kvinder og kan ramme alle etniske grupper. OI opstår i livmoderen, og der findes ingen kur. Det opdages normalt, når en person brækker mange knogler; de kan få foretaget DNA-test for at kontrollere for OI. Det forekommer i 1 ud af 20.000 fødsler.
Spørgsmål og svar
Q: Hvad er osteogenesis imperfecta?
A: Osteogenesis imperfecta er en genetisk lidelse, der almindeligvis kaldes knogleskørhed. Den svækker eller ødelægger kollagenstaven, som giver knoglerne styrke og fører til knogler, der er mere tilbøjelige til at brække.
Q: Hvordan nedarves osteogenesis imperfecta?
A: Osteogenesis imperfecta er normalt en autosomal dominant sygdom, hvilket betyder, at en person kan få den, hvis kun en af deres forældre har det unormale gen.
Q: Hvem identificerede først osteogenesis imperfecta?
A: Vrolik identificerede først osteogenesis imperfecta i 1849.
Q: Findes der en kur mod osteogenesis imperfecta?
A: Desværre findes der ingen kur mod osteogenesis imperfecta.
Q: Hvad er de fire typer af osteogenesis imperfecta?
A: De fire typer af osteogenesis imperfecta er type et, type to, type tre og type fire.
Q: Hvad er symptomerne på osteogenesis imperfecta type tre?
A: Personer med type tre af osteogenesis imperfecta kan have mere end 100 frakturer før puberteten. Deres øjne får ofte et lilla, blåt eller gråt skær, og folk med denne type har også ofte høretab.
Q: Hvor almindeligt er høretab hos mennesker med osteogenesis imperfecta?
A: 50% af dem, der har osteogenesis imperfecta, har høretab, mens de er ved at blive voksne.
Relaterede artikler
Forfatter
AlegsaOnline.com Skør knoglesygdom (Osteogenesis imperfecta) – årsager, typer og symptomer Leandro Alegsa
URL: https://da.alegsaonline.com/art/73422
Kilder
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"